

Welcome to QRS 2024 - 17th Quantum Reactive Scattering Workshop, hosted in the enchanting city of Istanbul.
On behalf of the Organizing Committee, we extend a heartfelt welcome to all participants. As we gather in Istanbul,
a city renowned for its rich history and the captivating fusion of Asia and Europe,
we invite you to embark on a journey that seamlessly blends academic excellence with cultural immersion.
QRS 2024 serves as a robust platform for exploring diverse facets of computational chemistry.
It continues the tradition of academic excellence initiated by David Clary in 1990 while adding a new chapter
to its global journey.
Now, for the first time, this esteemed event is poised to grace Türkiye, infusing new geographical and cultural dimensions into its distinguished legacy.
The Yıldız Campus, our chosen venue,
stands as an integral part of the illustrious Yıldız Palace,
acknowledged as a UNESCO World Heritage site.
With its origins dating back to 1880, the Yıldız Palace assumed a central role in
Türkiye's history,
particularly between 1876 and 1908, serving as a key administrative hub following the iconic
Topkapı Palace and
Dolmabahçe Palace.
Our profound gratitude is extended to the Yıldız Technical University Rectorate
for their cordial hospitality and exceptional stewardship in hosting QRS 2024.
We eagerly look forward to your participation in QRS 2024, to be held in the vibrant city of Istanbul.
Join us for an unforgettable QRS experience, marked by pioneering scientific discussions, productive collaborations, and captivating social events.
DATE 24 - 28 June 2024
LOCATION YTU, Beşiktaş, Istanbul
SPEAKERS
56 invited speakers
from 🌍 16 countries
Powered by Alpine Dynamics
Contact QRS 2024 at qrs2024@gmail.com
Invited Speakers
(Confirmed)
🇯🇵 Japan:2 🇵🇱 Poland:3 🇵🇹 Portugal:1 🇪🇸 Spain:12 🇹🇳 Tunisia:1 🇬🇧 United Kingdom: 3 🇺🇸 United States:10

Satrajit Adhikari
Indian Association for the Cultivation of Science, INDIA


Dmitri Babikov
Marquette University, UNITED STATES

Luis Bañares
Universidad Complutense de Madrid, SPAIN


Laurent Bonnet
Bordeaux University, FRANCE

Joel Bowman
Emory University, UNITED STATES

David C. Clary
University of Oxford, UNITED KINGDOM

Cecilia Coletti
University of Chieti, ITALY

Gianpiero Colonna
Institute for Plasma Science & Technology, ITALY

Jonathan Connor
University of Manchester, UNITED KINGDOM

Marko T. Cvitaš
University of Zagreb, CROATIA

Gábor Czakó
University of Szeged, HUNGARY

Fabrizio Esposito
Institute for Plasma Science & Technology, ITALY


Hong Gao
Chinese Academy of Sciences, CHINA

Jesús González-Vázquez
Autonomous University of Madrid, SPAIN

Hua Guo
University of New Mexico, UNITED STATES

Majdi Hochlaf
Gustave Eiffel University, FRANCE

Pablo G. Jambrina
University of Salamanca, SPAIN


Dariusz Kędziera
Nicolaus Copernicus University, POLAND

Boutheina Kerkeni
Tunis El Manar University, TUNISIA

Stephen J. Klippenstein
Argonne National Laboratory, UNITED STATES

Viatcheslav Kokoouline
University of Central Florida, UNITED STATES

Anna I. Krylov
University of Southern California, UNITED STATES

Jochen Kupper
University of Hamburg, GERMANY

Vincenzo Laporta
National Research Council Bari, ITALY

Manuel Lara
TUAM Madrid, SPAIN

Annarita Laricchiuta
National Research Council Bari, ITALY

György Lendvay
Research Center for Natural Sciences, HUNGARY


François Lique
University of Rennes, FRANCE

Andrea Lombardi
University of Perugia, ITALY

Balakrishnan Naduvalath
University of Nevada, UNITED STATES

Andrew J. Orr-Ewing
University of Bristol, UNITED KINGDOM

Daniel Peláez
Université Paris Saclay, FRANCE



Peter Saalfrank
University of Potsdam, GERMANY

Steven D. Schwartz
University of Arizona, UNITED STATES

Wojtek Skomorowski
University of Warsaw, POLAND

Petr Slaviček
University of Chemistry and Technology, CZECHIA


Ignacio R. Sola
Complutense University of Madrid, SPAIN

Thierry Stoecklin
University of Bordeaux, FRANCE

Arthur G. Suits
University of Missouri, UNITED STATES

Toshiyuki Takayanagi
Saitama University, JAPAN

Timur V. Tscherbul
University of Nevada, UNITED STATES

António J.C. Varandas
University of Coimbra, PORTUGAL

Saulo A. Vázquez
Universidade de Santiago de Compostela, SPAIN

Roland Wester
University of Innsbruck, AUSTRIA



Dong Hui Zhang
Dalian Institute of Chemical Physics, CHINA

Piotr Żuchowski
Nicolaus Copernicus University, POLAND
Featured Speakers
- Duncan Bossion, IPR - Université de Rennes, France
Machine learning prediction of state-to-state rate constants for astrochemistry - Pablo del Mazo-Sevillano, Universidad Autónoma de Madrid, Spain
Dealing with the spurious long-range interactions in PIP-NNs potential energy surfaces - Josh Forer, Columbia Astrophysics Laboratory, United States
Low-energy dissociative recombination of interstellar cations - Bilel Mehnen, Nicolaus Copernicus University, Poland
Exploring the Excited States of the HPS+ Cation through Electronic and Spin-Rovibrational Spectroscopy - Huilin Pan, Southern University of Science and Technology, China
Quantum interference induced by Fermi interactions in Cl + CH3D(v1-I, v1-II) → CH2D(00) + HCl(v=0, 1) - Hailin Zhao, Chinese Academy of Science, China
Theoretical Development of the Interaction-asymptotic Region Decomposition Method for Tetratomic Reactive Scattering
Poster Presentations
- Fatıma Büşra Aslan, Gebze Technical University, Türkiye
Periodic DFT investigation of CO hydrogenation to methanol on Zn/Cu(111) and Zn/Cu(211) surfaces with dispersion and Hubbard effects - Gökberk Çelikel, Gebze Technical University, Türkiye
Quantum Chemical Investigation of Methanol Decomposition on Copper Embedded Boron Nitride Surface to be Used in a Fuel Cell - Hamza Hendaoui, University of Tunis El Manar, Tunisia
Rotational excitation and de-excitation of interstellar chloronium cation in collisions with helium atoms - Mehmet Hanifi Kebiroğlu, Malatya Turgut Özal University, Türkiye
Enhancing the electronic properties of 2-Acetoxybenzoic acid through potassium doping: synthesis and characterization - Dora Papp, University of Szeged, Hungary
Modelling the dynamics of abstraction and substitution processes in polyatomic reactions - Marcin Stachowiak, Nicolaus Copernicus University in Toruń, Poland
Theory cracks old data: Rovibrational energy levels of orthoH2–CO derived from experiment - Yajian Shu, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, China
The Calculation of Geometric Phase Effects in the H+H2 Reaction within the Quantum Transition State Framework - Dicle Sevilmiş, Gebze Technical University, Türkiye
From CO to CH3OH on Cu (100) and Cu (110) Catalytic Surfaces with Hubbard and Dispersion Effects - Viktor Tajti, University of Szeged, Hungary
Theoretical characterization of the mechanisms of ion-molecule reactions - Petra Toth, University of Szeged, Hungary
Benchmark ab initio characterization, automated potential energy surface development and quasi-classical dynamics for the Cl + CH3CN reaction - Ransheng Wang, Chinese Academy of Science, China
Non-adiabatic effect in F/Cl atom plus hydrogen molecule and its deuterium isotopologue reaction - Yurun Xie, Institute of Advanced Science Facilities, Shenzhen, China
Preparation of an enhanced vibrational excitation hydrogen molecular beam for quantum scattering researches - Mücahit Yılmaz, Fırat University, Türkiye
Investigating the impact of sodium doping on 2-Acetoxybenzoic acid: synthesis and comprehensive characterization
Other participants
- Jie Han, Southern University of Science and Technology, China
- Oluş Özbek, Gebze Technical University, Türkiye
- Ezman Karabulut, Bitlis Eren University , Türkiye
- Mehmet Burak Aygün, Selçuk University, Türkiye
- Muhammed Kemal Çolak, Selçuk University, Türkiye
- Bensu Sıla Çelik, Anadolu University, Türkiye
- Gökalp Elaçmaz, Bilkent University, Türkiye
- Sevgi Sarıgul Ozbek, Acıbadem Mehmet Ali Aydınlar University, Türkiye
- Joanna Kozłowska, Nicolaus Copernicus University, Poland

Previous QRSs
The Quantum Reactive Scattering Workshop is a prestigious series of workshops initiated by David Clary in 1990. Over the years, this workshop has brought together luminaries and experts in the field from around the globe to foster collaboration.
- 1990 Cambridge, UK - David Clary
- 1994 Cambridge, Massachusetts, USA - Yan Sun and Michael Baer
- 1995 Nottingham, UK - David Clary and David Manolopoulos
- 1997 Telluride, Colorado, USA - Joel Bowman
- 1999 Perugia, Italy - Vincenzo Aquilanti and Antonio Laganà
- 2001 Pasadena, California, USA - Aron Kuppermann
- 2003 San Lorenzo de El Escorial, Spain - Javier Aoiz and Luis Bañares
- 2005 Santa Cruz, California, USA - Millard Alexander and Anne McCoy
- 2007 Cambridge, UK - Stuart Althorpe
- 2009 Dalian, China - Dong-Hui Zhang and Ke-Li Han
- 2011 Santa Fe, New Mexico, USA - Hua Guo
- 2013 Bordeaux, France - Laurent Bonnet and Pascal Larregaray
- 2015 Salamanca, Spain - Octavio Roncero, Tomás González-Lezana, Susana Gómez-Carrasco, Lola González-Sánchez
- 2017 Trieste, Italy - Niyazi Bulut, Noelia Faginas Lago, Andrea Lombardi, Federico Palazzetti
- 2019 Saitama, Japan - Toshiyuki Takayanagi
- 2022 Balatonföldvár, Hungary - György Lendvay, Gabriella Lendvayné Győrik, Ákos Bencsura
QRS 2024 Book of Abstracts

QRS 2024 Program
- 📅 Day 1 - 24 june 2024
- 📅 Day 2 - 25 june 2024
- 📅 Day 3 - 26 june 2024
- 📅 Day 4 - 27 june 2024
- 📅 Day 5 - 28 june 2024
- 📅 Day 1 - 24 June 202408:00 - 09:15 ✍ Registration
The registration desk will be cracking open from 08:00 until 12:30
09:15 - 09:30 David Clary Max Born and the Founders of Quantum Scattering TheoryThis talk will describe the very early development of quantum scattering theory with particular emphasis to the pioneering contributions of Max Born. Reference will be made to recent books by the speaker.
Session 1 - Chair Arthur Suits
09:30 - 10:05 Joel Bowman Progress in Developing Permutationally Invariant Machine Learned Potentials and Dynamics Using ThemI will review the dramatic progress in using permutationally invariant polynomials (PIPs) in machine-learned potential energy surfaces, the heart of our field. The high speed and high precision of the PIP approach, relative to a number of other methods, including ACE, GAP, ANI, sGDML, etc., will be shown for H3O2-, ethanol, glycine, and 21-atom aspirin. Very recent extensions to even larger molecules using the 'fragmentation' and 'n-body' methods will be presented as well. Applications to tunneling splitting in tropolone as well as diffusion Monte Carlo calculations of the zero-point energies of the eight low-lying conformers of glycine will be described. A brief overview of recent work on 'roaming' and old work on collision-induced isomerization will also be given.
10:05 - 10:35 Octavio Roncero Quantum Study of the CH3+ Photodissociation in Full Dimension Neural Networks Potential Energy SurfacesCH3+, a crucial intermediate in interstellar chemistry, was recently detected for the first time by the James Webb Space Telescope. This discovery has propelled studies into its photodissociation mechanisms. We have conducted accurate explicitly correlated multi-reference configuration interaction ab initio calculations and developed full-dimensional potential energy surfaces for the three lower electronic states using a fundamental invariant neural network method. This method accounts for the permutation symmetry of identical hydrogen atoms. The photodissociation cross section was calculated using a full-dimensional quantum wave packet method, employed in heliocentric Radau coordinates. The wave packet was represented on angular and radial grids, which helped to reduce the number of physically accessible points and manage spurious states that emerge when evaluating angular kinetic terms through a projection technique. The photodissociation spectra were calculated for several initial vibrational states of CH3+ in the ground electronic state, particularly those observed by the James Webb Space Telescope. Incorporating the new photodissociation cross sections into astrochemical models to simulate conditions in the Orion Bar indicates a lesser destruction of CH3+ compared to results obtained using recommended values from the kinetic database for astrochemistry (KIDA).
10:35 - 11:00 ☕️ Coffee BreakSession 2 - Chair Joel Bowman
11:00 - 11:35 Hua Guo Dynamics and Kinetics of Small Molecule Activation by Transition Metal/Oxide IonsReactions between transition metal/oxide ions and small molecules (H2, CH4, CO2, etc.) often require intersystem crossing between different spin manifolds. This is because the lowest spin-orbit state of the reactant typically has a high barrier, which makes the reaction infeasible without transitions to a different spin state with a low barrier. This so-called 'two-state reactivity' model has been invoked in rationalizing many ion-molecule reactions involving transition metals. In this talk, we present nonadiabatic simulations of multi-state reaction dynamics on machine learned global potential energy surfaces. These extensive studies help to elucidate key questions concerning the kinetics and dynamics of these important model systems for single-atom catalysis.
11:35 - 12:05 Satrajit Adhikari Role of Electron-Nuclear Coupling on Multi-State Reactive Scattering ProcessesThe first principle based beyond Born-Oppenheimer (BBO) theory is presented for the construction of highly accurate diabatic potential energy matrix to explore multi-mode multi-surface photoelectron spectra [1-4], reactive collision processes [5-10] and phase transition of solid [11-12]. The ab initio adiabatic potential energy surfaces (PESs) and nonadiabatic coupling terms (NACTs) are employed in order to calculate those diabatic surfaces. Jahn-Teller (JT) and Renner-Teller (RT) types of conical intersections (CIs) along with Pseudo Jahn-Teller (PJT) interactions and semi-circular CI seams are investigated for various spectroscopic systems [4], scattering processes [5-10] and perovskite species [11,12]. In this lecture, we primarily focus on reaction attributes (reaction probabilities, cross-sections and/or rate constants) of D+ + H2, He + H2+, F + H2 processes calculated by scattering dynamics over global diabatic surfaces of H3+, HeH2+, F + H2 systems, respectively, which exhibit good accord with the experimentally measured ones [8-10]. Finally, we show the effect of electron nuclear coupling in photoelectron spectra and ferroelectricity of TiO68- in the lattice, BaTiO3.
12:05 - 12:30 Pablo García Jambrina Stereodynamical Control of Inelastic Collisions at Low Energies: Three and Four Vector CorrelationsThe outcome of a bimolecular collision depends on the relative orientation and alignment of the collision partners before their interaction begins. In gas-phase bimolecular reactions, this can be addressed by polarizing the bond axis and/or rotational angular momentum of the reactants. Here we will illustrate how the outcome of a collision can be controlled by choosing the relative geometry of the colliding partners before they begin to interact. In particular, we will focus on inelastic collisions at low energies that proceed with contributions from only a few partial waves, such as those between two aligned D2 molecules whose angular distributions were recently measured by Zhou et al. [1]. Our results, based on full-dimensional coupled-channel scattering calculations, show that a higher degree of control can be achieved by the simultaneous alignment of two molecules, and that this control is more important when both partners change their internal state during the collision [2].
12:30 - 14:00 🍴 LunchSession 3 - Chair Hua Guo
14:00 - 14:35 Stephen Klippenstein Water Catalysis in Atmospheric ChemistryAt low temperatures, many atmospheric species have a significant probability of being in weakly bound complexes with water. The existence of such dimers can have dramatic effects on their chemical reactivity; changing both the rate constants and the product branching. We will use high-level ab initio transition state theory based master equation calculations to examine the effect of such complexes on the kinetics for three reactions: (i) the reaction of CH4 with OH, (ii) the reaction of CH2OO with H2O2, and (iii) the reaction of HO2 with HO2.
14:35 - 15:05 Steven Schwartz Quantum tunneling and evolution in enzymatically catalyzed reactionsWork in our group first identified the importance of femtosecond protein motions in the catalytic mechanism of enzymes. Through the analysis of sequences of lab evolved artificial enzymes we saw such motions absent in the original design build in as enzymes became more proficient. We now ask if similar evolutionary pressures might employ quantum tunnelling as an enzyme becomes more proficient. After a review of our promoting vibration concept in a variety of enzymes, we will present studies of proton transfer reactions in lab evolved artificial enzymes with both classical Transition Path Sampling methods and Path Integral Centroid methods to investigate the evolvable contributions of tunnelling.
15:05 - 15:25 Jonathan Connor A single resonance Regge pole dominates the forward-angle scattering of the state-to-state F + H2 → FH + H reaction at Etrans= 62.09 meV.An important and long-standing problem is: Does a resonance contribute to the forward-angle scattering of the F + H2 reaction? A well-defined, yet rigorous and unambiguous, investigation is made of structure in the differential cross sections (DCSs) for three state-to-state reactions at a translational energy of 62.09 meV. Firstly, accurate numerical scattering matrix elements are obtained for the Fu-Xu-Zhang potential energy surface. Secondly, the following theoretical techniques are employed: (a) Full and Nearside-Farside (NF) partial wave series (PWS) and local angular momentum theory, including resummations up to second order. (b) The recently introduced 'CoroGlo' test, which lets us distinguish between glory and corona scattering at forward angles for a Legendre PWS. (c) Six asymptotic (semiclassical) forward-angle glory theories and three asymptotic farside rainbow theories, valid for rainbows at sideward-scattering angles. (d) Complex angular momentum theories of forward and backward scattering, with the Regge pole positions and residues computed by Thiele Rational Interpolation. Thirdly, we conclude: (a) The forward-angle peaks arise from glory scattering. (b) There is a broad (hidden) farside rainbow at sideward angles. (c) A single Regge pole contributes to the DCS across the whole angular range, being most prominent at forward angles. This proves that a resonance contributes to the DCSs for the three transitions. (d) The diffraction oscillations in the DCSs arise from NF interference between the Regge pole and direct subamplitudes.
15:25 - 15:50 Dmitri Sokolovski Energy dependence of reactive differential cross sections of the state-to-state F+H2 → FH+H reaction in the 62.07-101.67 meV range: oscillations, resonances and polesRecently, the structures appearing in small angle-differential cross sections (DCS) of the title reaction were studied in detail for transitions at a single translational energy Etrans=62.09 meV. A variety of semi-classical and complex angular momentum (CAM) techniques used in the analysis confirmed the structures to be interference patterns resulting from capture of the reactants into a single metastable state. We look at a bigger picture and analyse the behaviour of the three DCSs in a broader energy range, relying on a simple CAM approach. We show that interference patterns seen in the forward, backward, and sideway DCSs are largely caused by the presence of a single dominant resonance pole for all energies considered in the study. We also find several weak narrow resonances, and in one case, an important Regge zero, which also leave their marks in the angular distributions.
15:50 - 16:15 ☕️ Coffee BreakSession 4 - Chair Tomás González Lezana
16:15 - 16:45 Laurent Bonnet Chemical Reaction Thresholds According to Classical-Limit Quantum DynamicsClassical-limit quantum dynamics will be used to explain the origin of the quantum thresholds of chemical reactions from their classical dynamics when these are vibrationally non-adiabatic across the interaction region. We will be working within the framework of an elementary model of chemical reaction that mimics the passage from the free rotation of the reagents to the bending vibration at the transition state to the free rotation of the products.
16:45 - 17:15 Dmitri Babikov Recent Progress in Development and Applications of Mixed Quantum/Classical Theory of Molecular CollisionsIn recent years significant progress has been made in the development and applications of an approximate theory for the description of molecule + molecule collisions. This theory rigorously treats the internal (rotational, vibrational) motion of collision partners using the time-dependent Schrödinger equation, while the translational motion (scattering) is handled classically via mean-field trajectories. This approach accurately predicts state-to-state transition cross sections at high collision energies and provides reasonable results down to the threshold for excitation of rotational and vibrational states. It captures major quantum effects such as symmetry of allowed and forbidden transitions, weak selection rules, propensity, and exchange symmetry of identical collision partners. It also addresses the issue of vibrational zero-point energy leakage. The framework allows for a hierarchy of approximations, making calculations more affordable and enabling the treatment of the largest molecules and most complex collision partners to date. The method is implemented in a user-ready suite of codes named MQCT 2024, available through GitHub (MarquetteQuantum). Examples include calculations for C6H6 + He, ND3 + D2, H2O + H2, and H2O + H2O, with ongoing work on rotational excitation and quenching of small organic molecules relevant to astrochemistry.
17:15 - 17:45 Cecilia Coletti The Many Faces of Vibrational Relaxation Dynamics in N2-O CollisionsThe knowledge of state-to-state rate coefficients for collisional vibrational excitation/relaxation in molecules is necessary for the modelling of non-local thermal equilibrium conditions, characterizing many gaseous and plasma environments in a variety of temperature ranges: interstellar media, planetary atmospheres, hypersonic flows, etc. In the last years we developed a computational protocol [1] capable of tackling often-neglected relevant issues: we use mixed quantum-classical methods, which explicitly account for quantum effects in the vibrational motion, and PESs accurately describing the long-range interaction regions. A new general formulation of the code, together with a Machine Learning implementation [2], enables to generate complete datasets for vibrational energy exchange processes. Among the investigated systems, vibrational relaxation in O+N2 collisions is particularly intricate and intriguing: O(3P)-N2 collisions occur on a manifold of spin-orbit states and only the inclusion of all the corresponding PESs and of their related processes allowed us to reconcile the long-existing discrepancies between experimental and calculated data [3]. The description of the O(1D)-N2 collisional dynamics results even richer and is complicated by the lack of experimental data to compare with: the process spans five PESs with (1Σ), (1Π) and (1Δ) character, the latter two -made of two asymptotically degenerate surfaces each- are non-reactive and strongly determine N2 vibrational distribution. The (1Σ) PES is instead characterized by several low lying N2O minima, whose lifetime can or cannot lead to efficient intravibrational redistribution. This determines the onset of a variety of highly stereodynamical energy-dependent vibrational excitation/de-excitation mechanisms, whose description requires the use of complementary theoretical approaches.
17:45 - 18:00 Huilin Pan Quantum interference induced by Fermi interactions in Cl + CH3D(v1-I, v1-II) → CH2D(00) + HCl(v0=0, 1)Mode-selectivity is a well-established concept in chemical dynamics. A polyatomic molecule possesses multiple vibrational modes; the mechanical couplings between them result in complicated anharmonic motions, defying a simple oscillatory description. Fermi-coupled vibration is a prototypical example, in which an energy-split eigenstate executes a coherent nuclei-motion comprising the characters of constituent normal modes with distinctive phases. Will this vibrational phase affect chemical reactivity? How to disentangle the phase effect from the amplitude effect? Here we report an experiment to address these questions in the reaction of Cl-atom with a pair of Fermi-states of CH3D(v1-I and v1-II). We found that the reactivity ratio of (v1-I)/(v1-II) in forming the CH2D(v = 0) + HCl(v) products deviates significantly from that permitted by the conventional reactivity-borrowing framework. Based on a proposed metric, this discrepancy can only be explained when the scattering interferences mediated by the CH3D vibrational phases are explicitly considered [1], which expands the concept of vibrational control of chemical reactivity into the quantum regime. In terms of more detailed state-to-state distributions, both the product state and the pair-correlated angular distributions in reactions with the two Fermi-paired CH3D(v1-I and v1-II) reactant states show striking similarities - only minor differences are noted. This "loss-of-memory" finding is counterintuitive. On the other hand, comparing to the corresponding dynamical attributes in the reaction with CH3D(v4=1), where v4=1 denotes one quantum excitation of the anti-symmetric stretching mode, distinct mode-specific behaviours are clearly revealed [2, 3]. The precise mechanisms underpinning those perplexing observations call for future theoretical investigations.
18:00 - 18:15 Hailin Zhao Theoretical Development of the Interaction-asymptotic Region Decomposition Method for Tetratomic Reactive ScatteringAn accurate and efficient time-dependent wave packet method is developed for solving the product state-resolved reaction probabilities of the tetratomic reactive system. This method is based on the previous interaction-asymptotic region decomposition (IARD) approach in the triatomic reactive system. By using this method, the entire scattering process is divided into the interaction region and multiple asymptotic regions. The hyperspherical coordinate can be applied in the interaction region, while the corresponding Jacobi coordinate can be employed in each asymptotic region. Therefore, the `coordinate problem' - the difficulty of expressing the wave function in the entire region using a single coordinate system - can be effectively resolved. Since the different optimal coordinates are adopted in different regions, a smaller number of grid points (or basis functions) are required to achieve convergent results. As a result, the IARD method is more efficient compared to previous wave packet methods. The typical tetratomic reaction H2 + OH with zero total angular momentum is studied using the IARD method and other wave packet methods. A comparative study can visually demonstrate the efficiency and accuracy of the IARD method. Furthermore, H2 (v=2) + OH reaction is studied accurately using the IARD method and the product with low translation energy appears.
18:15 - 18:25 🎻 Violinist Ozan Cem Baş Mini Classical Music Concert: BachOzan is a final-year student in the Faculty of Science at Bilkent University. He will delight our ears at QRS. Get ready for a captivating performance.
18:25 - 18:35 📷 QRS 2024 Photo SessionWe are inviting you, as participants of QRS 2024, for a group photo shoot at the designated area. Please, be part of the picture!
18:35 - 20:00 Reception starts with Hasan Mandal, the president of TUBITAK, giving a welcome speechThe president of TUBITAK (The Scientific and Technological Research Council of Turkey), Dr. Hasan Mandal, will commence the reception with an insightful talk, sharing his thoughts and vision for the advancements in science and technology in Türkiye.
-
📅 Day 2 - 25 june 2024
Session 1 - Chair Octavio Roncero
08:30 - 09:05 Anna Krylov States in the Continuum and Their Spectroscopic SignaturesOwing to the progress in many-body theories and computer hardware, quantum chemistry tools are now routinely used in chemistry and physics, providing both high-quality quantitative data (often rivaling the experimental measurements) and invaluable qualitative insights (crucial for interpretation of the experimental observations). Despite its success in treating a broad range of electronic structures, quantum chemistry is still lagging behind when electronically metastable states (i.e., resonances embedded in the ionization continuum) are concerned. This lecture will provide a concise overview of the progress in extending quantum chemistry into the continuum. The main emphasis will be on exploring the ideas of non-Hermitian quantum mechanics within the framework of coupled-cluster and equation-of-motion coupled-cluster theory. The performance of new theoretical tools will be illustrated by examples from electron scattering experiments.
09:05 - 09:35 Majdi Hochlaf Unimolecular Chemical Processes Upon (Multi)-Photon Ionization: Theory and ExperimentThe dissociation of doubly charged OCS2+ and SO22+ ions is examined. These doubly charged ions are formed by photoionization of the neutral molecule at 40.81 eV or by ultrafast multi-ionization processes. For interpretation, we use high-level quantum chemical calculations on isomeric structures and their potential energy surfaces. For SO22+, we find experimental evidence for electronic-state-selective production of O2+ from SO2, a chemical constituent of many planetary atmospheres and one which played an important part on Earth in the Great Oxidation Event. These experimental findings are analyzed with the aid of advanced ab initio computations resulting in the following model: First, the single photon double ionization of SO2 forms the SO22+ dication. The O2+ production involves isomerization of SO22+ by a roaming mechanism leading to efficient formation of the O2+ ion in the form of dissociative double ionization of SO2, which can be converted to abiotic O2 by electron neutralization. This formation process may contribute significantly to the abundance of O2 and related ions in planetary atmospheres, such as the Jovian moons Io, Europa, and Ganymede. It represents an alternative to and may compete with the well-established abiotic O2 production pathways via the photodissociation of water vapor by extreme ultraviolet (XUV) light [2-3] or the near ultraviolet (NUV) photochemistry of titanium (IV) oxide (titania) [4]. We suggest that this sort of ionic pathway for the formation of abiotic O2 involving multiply-charged molecular ion decomposition may also exist for other atmospheric and planetary molecules. Further details can be found in [5]. For OCS2+, the dominant dissociation channel of [OCS]2+ is charge separation forming CO+ + S+ ion pairs, found here to be formed with low intensity at a lower-energy onset and with a correspondingly smaller kinetic energy release than in the more intense higher energy channel previously reported. We explain the formation of CO+ + S+ ion pairs at low as well as higher ionization energies by the existence of two predissociation channels, one involving a newly identified COS2+ metastable state. We conclude that the dominant CO+ + S+ channel with 5.2 eV kinetic energy release is reached upon OCS2+ COS2+ isomerization, whereas the smaller kinetic energy release (of ~4 eV) results from the direct fragmentation of OCS2+ (X3Σ-) ions. Dissociation of the COS2+ isomer also explains the existence of the minor C+ + SO+ ion pair channel. We suggest that isomerization prior to dissociation may be a widespread mechanism in dications and more generally in multiply charged ion dissociations. Further details can be found in [6].
09:35 - 09:50 Josh Forer Low-Energy Dissociative Recombination of Interstellar CationsDissociative recombination (DR) of an electron colliding with a molecular cation, leading to the dissociation of the neutral system, is a major destruction pathway in molecular plasmas. DR plays an important role in the low-temperature, partially ionized star-forming gas clouds of the interstellar medium. Several interstellar molecular cations are key to constraining properties of diffuse interstellar clouds, allowing astronomers to better understand and model cloud collapse and ensuing star formation. Low-energy DR is of particular importance due to the low temperatures of diffuse interstellar clouds. However, low-energy DR has proven to be particularly challenging to study experimentally and theoretically, especially in the case of molecules with low-lying electronic resonances where the direct and indirect DR mechanisms may be simultaneously important. Only recently have storage ring experiments, carried out at the Cryogenic Storage Ring, been able to sufficiently cool the target ions so that ground-state DR cross sections can be measured. Theoretically, it is not known a priori which DR mechanism dominates, if either, and important dissociation pathways may not all have been identified for the target system. Here, we present recent developments in our theoretical DR studies of the following key interstellar ions: CH+, CF+, OH+, and H2O+. We make use of the UKRmol+ R-matrix electron-scattering codes, rovibrational frame transformation theory, and multichannel quantum defect theory to implicitly treat both direct and indirect mechanisms on even footing.
09:50 - 10:05 Duncan Bossion Machine Learning Prediction of State-to-State Rate Constants for AstrochemistryThe interpretation of astronomical observations requires the knowledge of the population of molecules in their rovibrational states. In the interstellar medium, local thermal equilibrium is not always established, and accurate knowledge of the reaction rate constants at the state-to-state level is essential. Calculations are often not feasible using only exact quantum methods, requiring datasets to be completed using more approximate methods. I will present how, by using an artificial neural network, it is possible to predict a large number of accurate state-to-state rate constants for atom-diatom collisions from available rates obtained at two different accuracy levels using a few accurate rates and many low-accuracy rates [1]. The H + H2 → H2 + H chemical reaction is used to benchmark the neural network, as both low and high accuracy state-to-state rates are available in the literature. The rates obtained are accurate even when using as low as 1% of the full dataset to train the neural network and significantly improve the low accuracy rates previously available. This approach can be used to generate full rate constant datasets with consistent accuracy from sparse rates obtained with various methods of different accuracies.
10:05 - 10:30 ☕️ Coffee BreakSession 2 - Chair Stephen Klippenstein
10:30 - 11:05 Dong Hui Zhang Accurate Potential Energy Surfaces and Quantum Dynamics for Polyatomic Chemical ReactionsIn this talk, I will present some of our recent works on quantum dynamics studies of some polyatomic reactions. Highly accurate potential energy surfaces have been constructed for these systems by using the Fundamental Invariant Neural Network (FI-NN) method based on extensive ab initio calculations. Time-dependent wave packet method has been employed to study these reactions at the state-to-state level. Dynamics resonances and the effects of vibrational excitation on the reactions will be discussed.
11:05 - 11:40 Timur Tscherbul A Quantum Algorithm for Multichannel Quantum Reactive ScatteringNumerically exact quantum (reactive) scattering simulations are limited to few-atom reactions due to the exponential scaling of computational complexity with the number of degrees of freedom. To address the curse of dimensionality problem, we developed a general-purpose quantum algorithm for solving time-independent quantum scattering problems on near-term (noisy intermediate-scale NISQ) quantum computers. The algorithm is based on the S-matrix version of the Kohn variational principle, in which the scattering S-matrix is calculated by inverting the Hamiltonian matrix expressed in the basis of square integrable functions. The computational bottleneck of the classical algorithm (large symmetric matrix inversion) is addressed using the variational quantum linear solver (VQLS), a NISQ algorithm for solving systems of linear equations. We apply our algorithm to single- and multichannel quantum scattering problems and show how it can be scaled up to simulate the quantum collision dynamics of large polyatomic molecules which are currently beyond the reach of classical algorithms.
11:40 - 12:10 Gábor Czakó First-Principles Reaction Dynamics from 5 to 12 AtomsWe introduce a first-principles technique to describe the dynamics of polyatomic chemical reactions using high-level ab initio potential energy surfaces (PESs) and the quasi-classical trajectory method. To move towards a black-box type, robust application of the above-mentioned methodology, we developed the ROBOSURFER program system [1] for automatic PES generation and the ManyHF method [2] for solving electronic structure issues frequently occurring in certain regions of the configuration space. We apply these methods to study the dynamics and mechanisms of reactions of fundamental molecules with atoms, ions, and radicals. We show the success of ROBOSURFER in the case of various systems involving 5 to 12 atoms. With the help of ROBOSURFER, we have already developed full-dimensional coupled-cluster-quality PESs for the F/Cl/OH + C2H6, HBr/HI + C2H5, F- + CH3Br, Cl- + CH3I, OH- + CH3F/CH3I, NH2- + CH3I, F-/OH- + CH3CH2Cl, F- + NH2Cl/PH2Cl/SiH3Cl, F/Cl + CH3NH2, Cl + CH3CN, NH3/SO2 + CH2OO, and OH + Gly reactions. Dynamics simulations on these PESs reveal unexpected reaction pathways like the multi-inversion in F- + NH2Cl [3] and oxide-ion substitution in OH- + CH3F [4], the competition between the different reaction channels and mechanisms, vibrational and rotational mode-specificity, post-reaction energy distributions as well as excellent agreement with experiments [5,6].
12:10 - 12:25 Pablo Del Mazo-Sevillano Dealing with the Spurious Long-Range Interactions in PIP-NNs Potential Energy SurfacesPermutationally invariant polynomial neural networks (PIP-NNs) are one of the most popular machine learning methods for representing (reactive) potential energy surfaces. They provide a flexible framework with the ability to accurately fit complex energy landscapes while remaining computationally affordable. They can readily be used to describe the short-range interactions between intermolecular fragments but extra care must be taken when including the long-range interactions. Due to the non-linear nature of the neural networks, there will always be a spurious interaction between distant fragments, resulting in a non-physical energy transfer between them. In this talk, we discuss the nature of this non-physical long-range interaction and different ways of dealing with it. This will be illustrated by two reactive processes: H2CO + OH → HCO + H2O and H2 + H2+ → H3+ + H. We will show how to generate an accurate PES without long-range spurious interactions and discuss the dynamical results.
12:25 - 13:55 🍴 Lunch14:00 - 18:30 Free Time18:30-21:00 🍹Cocktail on a Boat: 🚢 Bosphorus Tour 🐟Let's embark on a mesmerizing journey to the Istanbul Bosphorus, where the continents of Asia and Europe converge in an enchanting embrace. Experience the epitome of luxury with the Bosphorus Tour: Savor delightful beverages, and indulge in tempting snacks, all while surrounded by breathtaking views aboard the Bosphorus Tour Ferry! Prepare to be captivated and pampered as you witness the beauty of the Bosphorus Strait. This is a truly unforgettable experience that will leave you with cherished memories of this magical voyage!
-
📅 Day 3 - 26 June 2024
Session 1 - Chair Dong Hui Zhang
08:30 - 09:05 Andrew Orr-Ewing Ultrafast photochemical dynamics of nitrophenol compounds in aqueous solutionBrown carbon aerosol in Earth's atmosphere absorbs solar radiation at visible wavelengths and contributes to the overall warming of the troposphere. It is released directly to the atmosphere by biomass burning, whether from wild fires, domestic fuels, or agricultural practices, and it degrades air quality. Brown carbon aerosol particles contain a complex matrix of organic constituents, of which the most significant contributors to absorption of near-UV and visible light are nitroaromatic compounds such as nitrophenols and nitrocatechols. These nitroaromatic compounds are persistent pollutants in the environment, with low quantum yields for solar-UV photolysis despite their strong π*←π absorption bands. Our recent experimental and computational studies have unraveled the near-UV photoinduced dynamics of aqueous solutions of nitrobenzene and 4-nitrophenol. Using transient absorption spectroscopy over femtosecond to microsecond timescales, multiple steps in the photochemical pathways are revealed. For example, in 4-nitrophenol, ultrafast internal conversion from the (diabatic) 1ππ* state to the 1nπ* state occurs within ~150 fs, and is followed by competitive internal conversion to the S0 state and intersystem crossing to the triplet manifold of electronic states. In the T1 state, the photoexcited molecules deprotonate, giving an additional pathway for relaxation back to the parent molecule on pH-dependent timescales of tens to hundreds of nanoseconds. Drawing on our experimental and computational data, mechanistic comparisons will be made between the near-UV photodynamics of 4-nitrophenol, its isomer 2-nitrophenol, 4-nitrocatechol, and 2,4-dinitrophenol. Our deductions about the photochemical pathways account for the low quantum yields for sunlight-induced photodegradation of these compounds in aqueous solution.
09:05 - 09:35 Arthur Suits State-to-State Scattering Dynamics of Vibrationally Excited NO Over Five Orders of Magnitude in Collision EnergyWe present differential cross sections for rotationally inelastic and spin-orbit changing collisions of NO, prepared in specific rotational levels in v=9 and 10, with rare gases. The method employs stimulated emission pumping to prepare initial states at high vibrational excitation, coupled with velocity map imaging to record the scattered products state selectively. These experiments are conducted in a unique instrument that allows for intrabeam scattering at collision energies below 0.05 cm-1, as well as a near-counterpropagating geometry that facilitates access to collision energies over 11,000 cm-1, with complete tunability in between. The differential cross sections obtained are compared to results from quantum close coupling calculations where available, providing stringent tests of theoretical models under extreme conditions.
09:35 - 10:05 François Lique The Excitation of C(3Pj) in the Interstellar MediumAccurate determination of physical conditions of interstellar clouds is a crucial step to better understand the life cycle of interstellar matter and the formation of stars and planets. A key parameter for determining these conditions from interstellar spectra is the calculation of accurate collisional rate coefficients of interstellar species with the most abundant species. Among the interstellar species, atomic carbon in its ground electronic state C(3Pj) occupies a key place as it is a precursor for the synthesis of many carbon-rich molecules and as an indicator of giant molecular cloud formation processes in the interstellar medium. Furthermore, C(3Pj) is an important coolant of the "cold" interstellar medium. Here, we study collisions between atomic carbon C(3Pj) with Helium (He) and molecular hydrogen (H2) at low collision energies. State-to-state integral and differential cross sections are theoretically derived from highly accurate quantum calculations and are compared to those obtained experimentally using crossed-beam experiments. We generally observe a very good agreement between experimental and theoretical data that demonstrates our ability to model nonadiabatic dynamics. However, discrepancies were found at specific collision energies for collisions with H2, which most likely originate from an incorrectly predicted quasi-bound state. These observations provide exciting prospects for further high-precision and low-energy investigations of scattering processes. New rate coefficients at temperatures relevant to astrochemical modeling are also provided. They will allow for an accurate estimation of C(3Pj) abundance in interstellar clouds and will provide better constraints for astrochemical models.
10:05 - 10:30 ☕️ Coffee BreakSession 2 - Chair Andrew Orr-Ewing
10:30 - 11:05 Toshiyuki Takayanagi Quantum Dynamics Calculations for Positron-Molecule ScatteringPositrons, the antiparticles of electrons, have long been utilized in various applications such as vacancy detection in materials and cancer detection in the human body. However, the detailed interaction between positrons and molecules has not been thoroughly understood at the atomic level. Recent experimental studies using high-resolution positron beams have revealed that many molecules can bind positrons, as evidenced by vibrationally resolved annihilation spectra [1]. Motivated by these experimental findings, we have initiated theoretical studies on positron-molecule scattering processes [2-10]. We have developed a practical method for calculating positron binding energies on the basis of gradient-corrected density-functional potential and vibrationally resolved annihilation spectra of molecules using Fermi-Golden rule resonance widths. In my presentation, I will briefly discuss our recent achievements in this project, where various computational techniques commonly employed in the field of quantum reactive scattering have been utilized.
11:05 - 11:35 Ignacio Sola Control of Photodissociation Reactions from the Initial StateThe manipulation of quantum interference is pivotal in coherent control, extensively utilized in applications such as quantum-state preparation, photochemical reactions, and quantum information. A fundamental aspect of quantum control in chemistry involves manipulating chemical reactions to maximize yields and enhance selectivity among potential fragmentation channels. Typically, this control paradigm involves a two-step process: initially, a pump pulse ignites the dynamics, creating a vibronic wave packet in the excited states. Subsequently, a control pulse guides the wave packet along the reaction coordinate, adjusting phases or inducing transitions between electronic states as necessary. Often, both the pump and control pulses are integrated into a single pulse, refined through pulse shapers and learning algorithms to achieve desired outcomes. In this study, we introduce a novel paradigm termed geometrical optimization, wherein control is exerted by optimizing the initial wave function. This approach essentially reverses the traditional sequence by applying a control pulse prior to the pump pulse. The initial coherences established are instrumental in dictating the reaction outcomes and selecting fragmentation channels in polyatomic molecules, especially under strong nonadiabatic conditions. We demonstrate this method's efficacy on the A band of CH3I, highlighting its potential in precise molecular fragmentation control.
11:35 - 12:05 Jochen Küpper Dynamics of Microsolvated (Bio)MoleculesObserving molecules in action through the recording of 'molecular movies', i.e., their spatiotemporal evolution during chemical dynamics, with atomic spatial and temporal resolution promises to revolutionize our understanding of the molecular sciences and to provide a time-dependent basis of chemistry. Experimentally, we build upon our approaches to prepare highly controlled samples that enable advanced imaging methods of individual molecular species directly in the molecular frame. We prepare highly controlled molecular samples for advanced ultrafast imaging experiments. This includes the preparation of ensembles of individual molecular species, e.g., single microsolvation environments, single conformers, or even single quantum states. Furthermore, the generated very cold samples are ideally suited to fix the molecules in space in laser-alignment or mixed-field orientation approaches. Here, we used the electric deflector to spatially separate different molecular species in combination with pump-probe velocity-map-imaging experiments, including utilization of 4D 'cameras'. We demonstrated that this powerful experimental approach reveals intimate details, e.g., on the radiation damage and fragmentation of water-water and pyrrole-water dimers as well as on the UV-induced dynamics in the near-UV-absorbing prototypical biomolecular indole-water system. We determined the time-dependent appearance of the different reaction products and disentangled the occurring ultrafast processes. This novel approach ensures that the reactants are well-known and that detailed characteristics of the specific reaction products are accessible – moving further toward the complete chemical-reactivity experiment.
12:05 - 13:40 🍴 LunchSession 3 - Chair Laurent Bonnet
13:40 - 14:15 Balakrishnan Naduvalath Stereodynamics of Cold Molecular CollisionsAdvances in quantum state preparations combined with molecular cooling and trapping technologies have enabled unprecedented control of molecular collision dynamics. This progress, achieved over the last two decades, has dramatically improved our understanding of molecular phenomena in the extreme quantum regime characterized by translational temperatures well below a kelvin. In this regime, collision outcomes are dominated by isolated partial waves, quantum threshold and quantum statistics effects, tiny energy splitting at the spin and hyperfine levels, and long-range forces. Collision outcomes are influenced not only by the quantum state preparation of the initial molecular states but also by the polarization of their rotational angular momentum, i.e., stereodynamics of molecular collisions. In this talk, I will provide an overview of recent theoretical developments for the description of stereodynamics of cold molecular collisions and their implications to cold controlled chemistry.
14:15 - 14:45 Tomás González Lezana From Reactive Scattering to Helium Droplets: Dynamics and SimulationsIn the last few years, we have investigated a series of atom-diatom reactions of astrophysical interest. Employing the complex-forming mechanisms governing the dynamics of these processes, we utilized a statistical quantum method to derive cross sections and rate constants for cosmological models. We will discuss examples such as H+ + H2 and its isotopic variants, alongside processes involving rare gas atoms and protons. Recent collaborative results with experimental groups on doped helium clusters are also presented, highlighting observations and characterizations of large ordered structures formed by helium atoms surrounding multiply charged ions, like Ca2+. These structures and other similar systems aim to enhance our understanding of ion solvation processes.
14:45 - 15:15 György Lendvay Curious Features of the Dynamics of the CH3 + HBr → CH4 + Br ReactionQuasiclassical trajectory calculations were performed to explore the correlation between potential energy surface (PES) and reaction dynamics for cases when there is a pre-reaction van der Waals well and a submerged potential barrier on the PES. Excitation functions typical to capture were obtained: Reaction cross sections diverge when the collision energy is reduced towards zero. However, the magnitude of the cross sections is larger than expected for such weak interaction. Vibrational excitation of the breaking bond further accelerates the reaction, although the potential barrier is early. We found that, when at the time of the encounter, the H-Br bond is stretched, covalent attraction arises between the partially free H atom and the CH3 reactant, and this causes enormous reactivity enhancement already when the H-Br bond is in the vib-rotational ground state. The calculations also revealed that capture, i.e., when the system enters the van der Waals well, does not guarantee reaction. Furthermore, in up to 50% of collisions that pass the potential barrier (the system visits regions of configuration space where the energy is below the reactant van der Waals minimum), no reaction occurs. Multiple crossing of any reasonable transition state dividing surface is also observed. The activation energy of the reaction is found to change from about -2 kJ/mol at 200 K to about over +10 kJ/mol at 1000 K, in agreement with recent experiments. Consequently, the activation energy of the reverse, endothermic reaction also should change from about 70 to about 90 kJ/mol in this temperature range.
15:15 - 15:40 Gianpiero Colonna The Need of Detailed Kinetic Schemes for the Plasma and AerospaceThermal and chemical non-equilibrium, a common characteristic in gas discharges and high enthalpy flows, appears when a large amount of energy is supplied to a gaseous system with a rate comparable with internal and chemical relaxation characteristic times. To model these effects, the state-to-state (StS) [1] approach is the most accurate, determining, at the same time, the evolution of the composition and of internal distributions, the latter obtained without assuming a specific functional form, contrarily to the multi-temperature approach. Due to the complexity of the StS kinetic scheme and to the lack of data, the so-called ground models [2] are commonly used, that, assuming negligible the internal excitation, only processes starting from the ground state of chemical species are considered, limiting the contribution of excited states to a selected number of interactions. However, this approximation can result in misleading conclusions. As an example, the role of complete sets of vibrationally-resolved electron-impact cross sections have been investigated for gas discharges in diatomic molecule mixtures [3] (N2, O2, H2), showing relevant effects in the species evolution. The synergy among chemical processes, internal relaxation and electron energy distribution increases the relevance of vibrational and electronically excited states. In this work some examples of the influence of accurate StS chemical schemes in gas discharges and in high enthalpy flows [4] are presented, showing the importance of using complete datasets in modeling non-equilibrium conditions.
15:40 - 16:05 ☕️ Coffee BreakSession 4 - Chair Majdi Hochlaf
16:05 - 16:35 Thierry Stoecklin Bending Relaxation of H2O by Collisions with Para- and Ortho-H2For many years, astrochemical models of the Interstellar Medium (ISM) only took into account the rotation of the detected molecules as a result of the cold temperatures (<100 K) prevailing in interstellar clouds. However, the study of warmer environments, such as star-forming regions or the envelope and atmospheres of evolved stars, has led to the detection of some of the most abundant polyatomic molecules in the ISM, such as HCN [1], C3 [2], and H2O [3], in highly excited vibrational levels. Rate coefficients for the collisions of these vibrationally excited molecules with the most abundant element in the ISM, H2, are then needed to model these environments. To address this need, the Close Coupling (CC) method for a linear rotor (H2) colliding with a rigid asymmetric top molecule (H2O) presented by Phillips et al. [4] is extended by including the coupling of rotation with all the vibrational modes of the water molecule. A new code [5] is developed allowing both to calculate the ro-vibrational H2O bound states and to solve the space-fixed CC equations using a log derivative propagator. It is here applied to the collision of para-H2O and ortho-H2O with the two spin modifications of H2 for several initial states of the water molecules. The calculated H2O rovibrational bound states and the calculated H2O bending relaxation rate at 295 K are found to be in good agreement with experimental results while the bending relaxation process is shown to be controlled by near-resonant energy transfer between H2O and H2. We eventually investigated the decoupling between bending and rotation and found that the use of such an approximation leads to errors ranging from 50% to 100% respectively for the bending relaxation and state-to-state cross sections.
16:35 - 17:05 Petr Slavíček Diabatization Strategies in Computational PhotodynamicsAb Initio computational photodynamics is gradually becoming a mature field, allowing us to model dynamical processes in the excited states for medium-sized molecules. While different strategies were developed to account for the non-adiabatic transitions, it turns out that the quality of the simulations is in most cases controlled by the electronic structure methods used and we can safely use even simple [1], pragmatic approaches to treat the non-adiabatic transitions [2]. The electronic structure toolbox for computational photodynamics is rather limited. The high-quality methods such as XMS-CASPT2 method enable us to study only molecules of comparably small size [3] and we need to restrict our focus on several low-lying states. This is difficult once we are interested in the high-energy states [4] such as those encountered in radiation chemistry. In my talk, I will discuss possible applications of diabatization strategies for on-the-fly simulations of different states. I will discuss the proton transfer reaction in doubly charged pyrrole-water complex, new types of coupled energy and proton transfer [5], and intermolecular Coulombic decay processes. I will also briefly discuss the possible machine learning approaches to diabatization [6]. If time permits, I will briefly discuss a completely different strategy based on the electronic force field.
17:05 - 17:30 Manuel Lara Cold and Ultracold Dynamics of the Collision D+ + H2 → DH + H+The reactive collision D+ + H2 → DH + H+ plays a significant role in the chemistry of the early universe and is one of the main sources of HD in interstellar clouds. Including an ion, the dynamics involves interactions of very long range, which require special methodological tools when approaching the collision threshold. In the first part of this project, already published [1, 2], we focused on the collision of deuterons with para-H2 (j=0). The R-4 character of the interaction required significant methodological improvements in the time-independent code, based in hyperspherical coordinates, used in the calculations. Propagations up to a hyper-radius of 105 a0 were required to converge the scattering length. In this second part, we are coping with a more exigent case: the collision with ortho-H2 (j=1). Due to the R-3 character of the quadrupolar interaction, the behavior turns anomalous at threshold (l=0), and the propagations should be extended up to millions of atomic units. Instead, in order to capture the nature of the singularity (while limiting the numerical effort) our strategy consists in: (i) performing an exact calculation up to a finite distance (beyond which the potential is 'weak'), and (ii) dealing with the external region using perturbation theory: a multichannel generalization of the perturbative approach in Ref [3] is used.
17:30 - 17:55 Andrea Lombardi Endohedral Fullerenes as Reaction Sites for Space ChemistryThe discovery of both neutral and cationic C60 in space [1] has inspired the idea that fullerene nanocages could form a protected site for non-conventional elementary chemical reaction pathways to occur under the typical conditions of the interstellar medium. This speculation, which has only partially been explored so far, but without specific application to astrochemistry [2,3], could indicate new routes to molecule formation in space, widening the role of carbon nanostructures. In the present contribution, this idea is briefly discussed, and some preliminary structure and dynamics calculations are presented for the H + C60 system.
17:55 - 18:10 Bilel Mehnen Exploring the Excited States of the HPS+ Cation through Electronic and Spin-Rovibrational SpectroscopyUsing post Hartree-Fock multi-configuration interaction ab initio approaches, the six (four) doublet (quartet) lowest electronic states of thioxophosphane cation, HPS+, are treated. These electronic states are mapped along the internal coordinates, including the HP, PS distances and in-plane HPS bending angle. Several potential minima are located where long-lived HPS+ ions can be found. Also, computations show that the (̃X2A', 12A'', 22A'', 12A', 14A' and 14A'') states are bound, for which we generate their 3D-potential energy surfaces (3D-PESs). After nuclear motion treatments, we determine the rotational and vibrational constants of these states and the pattern of their rovibrational levels up to 4000 cm-1 above the corresponding vibrational ground state level. For instance, the RCCSD(T)/aug-cc-pV(5+d)Z anharmonic frequencies for HPS+(̃X2A') are computed ν1 = 2237.7, ν2 = 543.0, ν3 = 686.8 (in cm-1). For the lowest electronic excited state (HPS+(12A'')), these frequencies change to ν1 = 2330.5, ν2 = 254.6 and ν3 = 749.6 (in cm-1). Several anharmonic resonances are identified. Besides, a very accurate adiabatic ionization energy of HPS (= 9.3173 eV) is deduced using the (U)CCSDT(Q)/aug-cc-pV(T+d)Z approach and where the core-valence (ΔCV), scalar relativistic (ΔSR) and zero point vibrational energy (ΔZPE) corrections are included. Our spectroscopic data should be useful for identifying this ion in laboratory and in media where reactive collisions between PS/PS+ + H+/H or HP/HP+ + S+/S are taking place.
18:10 - 18:30 Discussion and Decision Meeting: Next QRSDuring this meeting, our focus will be on deliberating and ultimately determining both the venue and the individual(s) responsible for orchestrating the upcoming QRS.
18:30 - 20:30 Poster SessionWe will be listening to 3-minute talks from poster presenters just before the poster session. The aim is to rev up our engines... But don't worry, there's no need for anyone to search for a restaurant for dinner. Beverages, snacks, and most importantly, gözleme will be available to enjoy during the discussions in front of the posters.
-
📅 Day 4 - 27 June 2024
Session 1 - Chair Toshiyuki Takayanagi
08:30 - 09:00 Luis Bañares Ultrafast Molecular Photodynamics Mediated by Conical IntersectionsIt is well accepted that the most general case of photoinduced reaction dynamics occurs through non-adiabatic transitions. The complex panorama of potential energy surfaces describing the excited states of polyatomic molecules is characterized by non-adiabatic crossings and the presence of multiple conical intersections. A conical intersection (CI) is a 3N-8 dimension hypersurface of intersection between two electronic states. The two remaining internal coordinates i.e. g as the difference gradient vector and h as the non-adiabatic coupling vector define the branching plane. Conical intersections can be considered as the transition states of electronic excited states and therefore the coupling between the different degrees of freedom valence electrons and vibrations and the timescales of these motions are at the heart of the understanding of photochemistry. The main aim is to find an equivalent of the `Polanyi rules' for excited state polyatomic dynamics in such a way that specific vibrational dynamics at CIs would be as important to dynamics as are the topographical features of the CIs themselves. In this contribution, we will highlight several cases of ultrafast non-adiabatic reaction dynamics in which CIs play a determining role for the photoinduced dynamics. In particular, we will focus on the non-adiabatic dynamics in vinyl iodide [1] and the methyl iodide cation [2].
09:00 - 09:30 Alberto García-Vela Quantum Control of Resonance Lifetimes in Molecular Photodissociation with Intense Laser FieldsA variety of molecular reaction processes are governed or mediated by resonance states. Some examples are the photodissociation of intermediate molecular species in atmospheric cycles, and the resonance-mediated reactive collisions of such intermediate species with other atmospheric reagents. When these resonance-mediated processes compete, control over the resonance behavior (essentially the resonance lifetime and the product fragment distributions) can be crucial in order to steer the molecular reaction process in a desired direction. In previous works, control schemes using weak laser fields were applied to induce quantum interference in order to modify the lifetime [1,2] and the product fragment distributions [3,4] of resonance-mediated molecular photodissociation. In the work presented here, moderately intense (of the order of \(10^{11}-10^{12} \, \text{W/cm}^2\)) laser fields are applied to change the resonance lifetime of a photodissociation process [5]. Several control schemes are suggested to reduce the lifetime of a long-lived resonance to make it as short-lived as desired. A control scheme able to increase the lifetime of a short-lived resonance will also be presented. The conditions required to apply the different schemes proposed will be discussed. The schemes appear to be of universal application to resonance-mediated molecular photodissociation.
09:30 - 10:00 Saulo Vázquez A Simple Approach to Improve the Accuracy of Semiempirical Methods for Intermolecular InteractionsIntermolecular (or noncovalent) interactions play a fundamental role in many chemical and physical processes. For large molecular systems and long-time dynamics, practical calculations are limited to either semiempirical quantum mechanical (SQM) methods or molecular mechanics force fields. Traditionally, the most popular SQM methods are those based on the neglect of diatomic differential overlap (NDDO) approximation (e.g., the PM6 method). The dramatic approximations involved in these methods lead to substantial errors in the calculation of intermolecular interactions and, consequently, different approaches have been proposed to correct them. More recently, the development of density functional tight binding (DFTB) has led to qualitative improvements in the accuracy of SQM methods. Nevertheless, for many systems and depending on the orientations of the interacting molecules, the accuracy of SQM methods is still unsatisfactory, and therefore further efforts are required to improve their performance. Here we show a simple approach to enhance the accuracy of SQM methods for noncovalent interactions. Specifically, we use analytical, pairwise corrections in which the parameters depend on the nature of the interacting functional groups. An example of the successfulness of the method is displayed below, where we compare interaction energies for 77 different complexes of the diglycine dimer, determined with the PM6 Hamiltonian, with our PM6-FGC method (FGC stands for functional group corrections) and, as a reference, with B3LYP-D3/def2-TZVP calculations.
10:00 - 10:25 ☕️ Coffee BreakSession 2 - Chair Luis Bañares
10:25 - 10:55 Roland Wester Ion-Molecule Reactive Scattering: From Classical to Quantum DynamicsCrossed-beam scattering experiments are a unique approach to study ion-molecule reaction dynamics [1,2]. In this talk I will present reactive scattering results for ion-molecule reactions that we have recently studied using our velocity map imaging spectrometer and where we have compared fluorinated with non-fluorinated ethyl halides [3]. Furthermore, an unexpected dynamical isotope effect has been observed in the reaction of fluorine anions with methyl iodide, F- + CH3I. The differential scattering data are compared with quasiclassical and quantum scattering calculations and the evidence for quantum dynamics in this reaction will be discussed [4]. Finally, results will be presented on the quantum dynamics in the proton transfer reaction D- + H2, which was studied in a cryogenic radiofrequency ion trap. At low temperature, this reaction proceeds via tunneling with an extremely small rate coefficient [5].
10:55 - 11:30 António Varandas From Molecular Structure to Dynamics: Attitudinal Green ApproachAb initio quantum chemistry is the rationale in molecular sciences. Although size is its hardest obstacle, it is also restrictive in establishing relationships between the parent molecule and the molecules (tiles) embedded on it. The sense of collective behaviour is then lost. Because ab initio calculations may attain near exactness for the tiles, this holds up quasi-molecule theory for the endeavour. This divide et impera strategy shows similarities to what is done in many-body expansion theory to construct global potential energy surfaces. Also at stake is the difficulty in using large basis sets to eagerly warrant variational accuracy. This is overcome by extrapolation of cost-effective energies and other properties to their complete basis set limit. A corresponding need holds for reaction quantum dynamics: while affordable at the quasi-classical level even for large molecules, the calculations are unbearable in quantum dynamics but for few-atom systems. Since both classical and quantum dynamics must merge asymptotically, the suggestion is that reliable quantum results obtained at a modest cost for zero-total angular momentum are extrapolated to their exact counterparts via extrapolation with quasiclassical ones. The message is thus that `attitudinal-red' brute-force calculations are replaced by `attitudinal-green' ones keeping accuracy at the horizon.
11:30 - 12:00 Dariusz Kedziera Generating the Potential Energy Surfaces for Complexes Relevant to AstrochemistryGenerating potential energy surfaces (PES) for complexes relevant to astrochemistry is a challenging task. Obtaining accurate PES for non-covalently bound molecular dimers presents two primary challenges. Firstly, there is the computational cost associated with accurately calculating interaction energies. Secondly, the number of grid points required increases exponentially with the dimensionality of the problem. One potential solution to the computational cost issue is to transition from established methods like CCSD(T) or CCSD(T)-F12 to SAPT or even more cost-effective SAPT(DFT). Both SAPT and SAPT(DFT) have demonstrated reliability in describing non-covalent interactions. Addressing the challenge of the exploding number of grid points can be achieved by seamlessly integrating short-range interactions (calculated within a given approximation) with long-range interactions (using multipole expansion). Additionally, rather than using evenly spaced grid points, selecting grid points based on physical information obtained from earlier versions of potentials for a given system can lead to significant savings in computational resources. These solutions have been implemented in the autoPES code developed in Szalewicz's group. This presentation will cover some technical aspects of the autoPES code, as well as successful applications such as the recently published studies on the benzonitrile-He [1] and HCN-H2O [2] systems.
12:00 - 12:25 Wojciech Skomorowski Towards Ab Initio Modelling of Penning Ionization ReactionPenning ionization is a reaction of the general form A* + M → A + M+ + e- where target atom or molecule M is ionized by collision with electronically excited reagent A*. Typically, A* is a noble-gas atom in its excited metastable state, such as He(3S) or Ne(3P). Due to its unique characteristics, Penning ionization has been the subject of significant experimental research using crossed-molecular beams, and, more recently, by means of novel merged beam technologies. Despite the importance of Penning ionization in multiple experimental settings and fundamental research, its first-principle modeling remains challenging due to the autoionizing nature of the electronic state in the entrance channel of the reaction. If coupling with the electronic continuum is to be included within the framework of the Born-Oppenheimer approximation, then complex-valued potential energy surfaces (CPES) are needed. In the talk, I will present a novel approach to calculate CPES for molecular systems undergoing Penning ionization. The method combines Fano-Feshbach resonance theory with equation-of-motion coupled-cluster (EOM-CCSD) wave function, and explicit description of the ejected electron. As numerical illustrations, I will show CPES for a few benchmark systems such as He(3S) + Ar, He(3S) + H2, or Ne(3P) + Ar. The reported CPES are compared with available empirical surfaces derived from the experimental data and with results of more sophisticated calculations based on explicit non-Hermitian quantum mechanics. The presented method with its simple and robust scheme opens up possibilities for fully ab initio quantum-dynamical studies of Penning ionization reaction, even for sizable molecular targets.
12:25 - 13:45 🍴 LunchSession 3 - Chair Oluş Özbek
13:45 - 14:10 Boutheina Kerkeni Reaction Paths of Complex Organic Molecules Formation on Ice Grains in the Universe: Quantum Chemical InvestigationsAstronomical observations have shown that carbonaceous compounds are ubiquitous in our and distant galaxies. Carbon is a key element in the evolution of prebiotic material (Henning and Salama 1998), and becomes biologically interesting in compounds with nitrogen, oxygen, and hydrogen. Interstellar molecular clouds and circumstellar envelopes are factories of complex molecular synthesis. For example, one of the most complex molecules: Propyl Cyanide (C3H7CN) observed in the interstellar medium has an isomer that shares the branched atomic backbone of amino acids. Another molecule: acetaldehyde (CH3CHO) is ubiquitous in interstellar space and is important for astrochemistry as it can contribute to the formation of amino acids through reaction with nitrogen-containing chemical species. The detection of formamide and acetamide is a proof of the existence of peptide bonds, --C--(O)--NH--, in the ISM of immediate interest in prebiotic chemistry. We provide quantum chemical calculations of reaction paths on water ice grains and emphasize the importance of introducing amounts of hydrons in the ice to catalyze the formation of amide bonds, which proves to dramatically reduce the reaction barriers, and transforms a bimolecular reaction into a unimolecular process. The study of the chemistry of Sulphur molecules in cold interstellar clouds is a very active area of research, especially since Sulphur is known to play an important role in biological systems. We investigate with ab initio methodologies the reaction processes involving the recently observed HCSCN and HCSCCH molecules, a project at the heart of the COST CA21126.
14:10 - 14:35 Fabrizio Esposito Cold Plasma Production of NO from Air: Motivation, Mechanism, Issues, and Possible SolutionsNitric oxide is commonly thought of as a pollutant coming from high temperature combustion in air. Indeed, it is a candidate for substituting the current reactive nitrogen industrial production with a completely green process [1], responsible, for example, for half of the world's food production by means of artificial fertilizers, but based on processes which are environmentally unsustainable. Unfortunately, nitric oxide production is economically unsustainable to date, due to the quite low energy efficiency in both industrial and laboratory realizations, which appears to be much lower than what one could expect on the basis of theoretical considerations involving the use of a non-equilibrium, cold plasma. Kinetic modeling could have a crucial role in guiding experimentalists in the search for an efficient solution, but the poor knowledge of some elementary processes [1] of fundamental importance in the mechanism behind NO production relegates this possibility to a dream for the future, at variance with compelling needs in the present. The molecular dynamics issues related to this problem will be presented [1-2], with some results recently achieved [3-4-5] and other possible solutions [1] requiring the use of more than one method to cover the wide total energy range explored by the kinetics involved.
14:35 - 15:00 Massimiliano Bartolomei Sodium Intercalation into γ-Graphyne Multilayers: A Theoretical Validation for Anodes in Metal-Ion BatteriesThe unfavorable intercalation of sodium (Na) ions into graphite layers has prevented the development of graphite-based anodes for Na-ion batteries with similar features to those commonly used in the ubiquitous lithium (Li)-ion batteries. In this contribution, we demonstrate that the recently synthesized γ-graphyne carbon material, a nanoporous derivative of graphene, can serve as an optimal host for Na ion intercalation. Each of its regular pores of sub-nanometric size features an 'ad hoc' environment for hosting a single Na ion, as reported here by means of high-level electronic structure calculations. We show that the graphyne/Na ion coupling mimics that found on graphene/Li ion in terms of both metal-single layer interaction and equilibrium distance. More importantly, unlike what is estimated for Na intercalation in graphite, we demonstrate that the binary intercalation compound formed with graphyne multilayers is thermodynamically stable. Moreover, such a compound mimics most of the features characterizing the anode material in Li-ion batteries in terms of storage capacity, structural properties, ion diffusion, and electrical conductivity, thus possibly paving the way for the development of novel Na-ion battery technologies.
15:00 - 15:20 Viatcheslav Kokouline Vibronic coupling in formation of negative molecular ionsSeveral negative molecular ions have been detected in the interstellar medium CN-, C3N-, C5N-, C7N-, C4H-, C6H-, C8H-, C10H- [1-5]. Since their discovery, for several years, there had been a general consensus in the community of modelers that in the interstellar medium (ISM) the ions are formed by the process of radiative electron attachment (REA). In a series of theoretical studies [6-11], we explored the REA process and the process inverse to it, photodetachment using ab initio methods. Cross sections and rate coefficients for formation of these ions by REA to the corresponding neutral radicals are calculated. For completeness of the theoretical approach, three pathways for the process have been considered: (i) A direct pathway, in which the electron in collision with the molecule spontaneously emits a photon and forms a negative ion in one of the lowest vibrational levels; (ii) an indirect, or two-step pathway, in which the electron is initially captured through non-Born-Oppenheimer coupling into a vibrationally resonant excited state of the anion, which then stabilizes by radiative decay; and (iii) a pathway via a weakly-bound dipolar state of the ion. The validity of our calculations is verified by comparing the present theoretical results with data from recent photodetachment experiments performed in Innsbruck. The second part of the study is devoted to an alternative process of formation of the negative molecular ions in the interstellar medium: by dissociative electron attachment (DEA). Ab initio studies were performed for DEA to H2CN and HNCCC, leading to formation of CN- and C3N- in the ISM. The results will be presented at the workshop.
15:20 - 15:45 ☕️ Coffee BreakSession 4 - Chair Roland Wester
15:45 - 16:10 Annarita Laricchiuta Simplified Approaches for Elementary Processes in Plasma KineticsThe kinetic modelling of non-equilibrium plasmas, relevant to different applications ranging from aerospace to technological discharges, requires accurate, complete and consistent datasets of cross sections and rate coefficients detailed on the internal degrees of freedom of molecular species. The methods in theoretical chemistry, enhanced by the modern techniques of high-performance computing, allow in principle the derivation of dynamical data in a quantum framework; however, still simplified/phenomenological approaches could be a valuable tool for the description, with a reasonable accuracy, of the excited species in the plasma. In this context, the threshold-modified Mott-Massey (TMMM) approximation and the Binary-Encounter-Bethe (BEB) approach [1,2] have been used for the derivation of vibrationally-resolved cross sections for excitation, dissociation and ionization of diatomic molecules induced by electron impact, while the Schwartz-Slawsky-Herzfeld (SSH) [3] and the semiclassical FHO (Forced Harmonic Oscillator) [4] methods are widely used in the community for the vibration to translation collisional energy transfer. Examples of the exploitation of these approaches in the completion of chemical schemes for the vibrational and electronic state kinetics of plasmas in aerospace applications will be discussed.
16:10 - 16:35 Lucia Daniela Pietanza Plasma-induced CO2 Dissociation: Self-consistent Coupling of a State-to-State Vibrational Kinetic Model with the Electron Boltzmann EquationIn the last decades, the increased emission of greenhouse gas and the decreasing reserves of traditional energy sources have renewed the interest in developing new technologies able to convert the CO2 captured from the atmosphere into useful chemical fuels, in a virtuous closed-loop carbon cycle. In this context, plasma-induced CO2 conversion under non-equilibrium conditions could offer an efficient and alternative route to conventional approaches for the CO2 dissociation into CO + O. Non-equilibrium plasma discharges can activate CO2 dissociation at low gas temperature through the selective excitation of vibrational levels by electron impact. This idea to exploit the vibrational-induced dissociation channel has received renewed interest by the CO2 plasma scientific community with the development of advanced kinetic simulation models and experiments [1]. In this contribution, we present the results obtained by a 0D State-to-State self-consistent kinetic model for the description of the plasma assisted CO2 dissociation in different types of plasma discharges, focusing on the crucial role of vibrational excitation [2-6]. The kinetic model is based on the simultaneous and self-consistent time-dependent solution of the equations describing the vibrational and electronic excited state kinetics and the plasma composition and the electron Boltzmann equation for the calculation of the electron energy distribution function. The reliability of the model is strongly dependent on the accuracy of the rate coefficients used. Discussion about the need of accurate and complete sets of state-specific rate constants for vibrational energy exchange in molecular collisions involving triatomic molecules, such as CO2, will be provided.
16:35 - 17:00 Vincenzo Laporta The Role of Electron-Molecule Collisions on Vibrational Relaxation of Non-Equilibrium PlasmaIn my presentation, I will illustrate my researches on non-equilibrium plasma modelling and on state-specific electron-molecule scattering. I will focus, in particular, on vibrational-excitation and dissociation (attachment, recombination, dissociative-excitation) collisional processes - rotationally and vibrationally resolved - involving neutral molecules or molecular ions with electrons and their role in the kinetic description of plasma out of equilibrium. The molecular electronic structures are obtained by using ab initio quantum chemistry approaches implemented in computer codes like MOLPRO and UK-RMatrix whereas the nuclear dynamics is studied within the theoretical models of Bardsley's local-complex-potential model, adiabatic-nuclei approximation and multichannel quantum defect theory. The latest results for cross sections and rate coefficients will be presented and discussed for NO [1], D2 [2], H2+ [3], ArH+ [4,5] and BeH+ [6,7]. Finally, I will present the results for the vibrational relaxation times [1,8,9] based on a master equation approach within a state-to-state scheme using the calculated cross sections.
17:45 🍴 Gala Dinner -
📅 Day 5 - 28 June 2024
Session 1- Chair Vincenzo Aquilanti
09:05 - 09:30 Manabu Kanno Electronic and Reaction Dynamics of Near-Infrared-Induced Conversion from Polyhydroxy Fullerenes to Graphene FlakesThe effect of chemical modification on the structural stability of C60 is an intriguing topic in nanocarbon chemistry. Krishna et al. experimentally examined the response of various C60 derivatives to the low-intensity (102 Wcm-2) near-infrared (785 nm) laser irradiation for several 10 seconds at room temperature [1]. Carbon nanotubes and carbon onions were produced only from the samples of polyhydroxy fullerenes (PHFs), indicating that the temperature of the system exceeded 2600 K. Krishna et al. explained the whole process of this reaction in the electronic ground state. However, even with low-intensity lasers, electronic excitation may still be possible due to long-time irradiation. To clarify this issue, we selected C60(OH)24 as a PHF molecule and investigated its Ehrenfest (mean-field) dynamics using the real-time time-dependent density-functional tight-binding (RT-TDDFTB) method [2]. Since a simulation on the order of 10 seconds is difficult, we assumed an intense (1012 Wcm-2) picosecond 785-nm laser pulse. The results of the RT-TDDFTB simulation showed a significant contribution of electronic excitation and suggested that C60(OH)24 is heated through electronic relaxation. In contrast, the energy of pristine C60 hardly increases upon irradiation with the same laser pulse. This means that hydroxy groups are responsible for the heating of PHFs. On the assumption of complete relaxation, we next calculated the reaction dynamics of heated C60(OH)24 (~ 2600 K) in the electronic ground state with the standard DFTB method. The hot PHF is so reactive that its C60 cage is broken into graphene flakes as a seed for various larger nanocarbons.
09:30 - 09:55 Hong Gao Collisional Charge-Transfer and Energy-Transfer Dynamics between Ar+(2P3/2,1/2) and N2In this work, we report a synergistic experimental and theoretical investigation on the collisional charge-transfer and energy-transfer dynamics between the spin-orbit state selected Ar+(2P3/2,1/2) ion and N2. The Ar+ ion is prepared in a specific spin-orbit level by using the resonance-enhanced multiphoton ionization method. The velocity and angular distributions of the charge-transfer product N2+ and the energy-transfer (inelastic) product Ar+ are measured at well-defined collision energies by using a home-built velocity-map imaging-based ion-molecule crossed-beam setup. Full-dimensional trajectory surface hopping (TSH) calculations are performed, which qualitatively reproduce the experimental measurements. For charge-transfer between Ar+ (2P3/2) and N2, product vibrational-state-specific mechanisms have been clearly revealed. Besides the well-known long range Harpoon mechanism for producing the predominant N2+(v'=1) channel, the hard collision glory mechanism recently identified in inelastic collisions is found to play an important role in producing the N2+(v'=2) channel, where highly rotationally excited products are scattered into the forward region, due to the delicate balance between attractive and repulsive forces of the collisional partners. For the charge transfer between Ar+ (2P1/2) and N2, different outcomes have been observed, where the product vibrational levels are partially resolved. The agreement between TSH calculation and experiment is not as good as that for Ar+ (2P3/2), further theoretical calculation is needed. For the collisional energy-transfer process, both experiment and TSH calculation show that all the vibrationally inelastically scattered N2 are forward peaked, contradicting the conventional wisdom for vibrational energy transfer processes. This is attributed to the hard collision glory mechanism recently identified in inelastic collisions between two neutrals.
09:55 - 10:20 Daniel Peláez Ruiz Analytical Low-Rank Sum-of-Products Representations of Bound and Unbound Local Operators in High-DimensionalityGrid-based representations are common to many and varied disciplines ranging from physics to computer science, as epitomised by quantum dynamical simulations or Deep Learning, respectively. Such type of problems require ordered and sizeable collections of data which can be formally viewed as multidimensional arrays or tensors. Handling these raw data-structures is difficult. Hence, accurate yet compact (low-rank) tensor approximations have become mandatory. In this respect, major advances in these fields have been linked to the adoption of functional forms for the efficient expression of multidimensional objects, such as the wavefunction [1]. In this contribution, we will discuss these aspects for the case of a smooth physical quantity. We will present our approaches for the re-expression of tensors into analytical low-rank of sum-of-products expansions using Tucker [2], Canonic Polyadic [3], and Hierarchical Tucker [4] ansätze. We will illustrate the application of these methods to bound systems (e.g., vibrational) [2, 3] as well as to unbound atom/molecule-surface scattering problems [5].
10:20 - 10:45 ☕️ Coffee BreakSession 2 - Chair António Varandas
10:45 - 11:15 Vincenzo Aquilanti Renormalized Chemical Kinetics and Benchmark Quantum Mechanical Rates: Activation Energies and Tunneling Transitivities for the F + H2/HD ReactionsExperimental, theoretical and computational chemical kinetics contributes to progress both in molecular and materials sciences and in biochemistry, exploring the gap between elementary processes and complex systems. Stationary state quantum mechanics and statistical thermodynamics provide interpretive tools and instruments for classical molecular dynamics simulations for stable or metastable structures and near-equilibrium situations. Chemical reaction kinetics plays a key role at the mesoscales: time-dependent and evolution problems are typically tackled phenomenologically, and reactions through intermediates and transition states need to be investigated and modeled. In this paper, scaling and renormalization procedures are developed beyond the Arrhenius equation and the Transition State Theory, concerning two key observables in reaction kinetics, the rate 'constant' as a function of temperature (and its reciprocal, the generalized lifetime), and the apparent activation energy (and its reciprocal, the transitivity function). Coupled first-order equations -- dependent on time and on temperature -- are formulated in alternative coupling representations: through a decoupling scheme, a link is provided from experimental results to effective modeling, or vice-versa from molecular dynamics simulations to predictions. The passage from thermal to tunneling regimes is uniformly treated and applied to converged quantum mechanical calculations of rate constants available for the prototypical three-atom reactions of fluorine atoms with both H2 and HD: these are exothermic processes dominated by moderate tunneling, needing formal extension to cover the low-temperature regime where aspects of universal behavior are shown to emerge. The results that have been validated towards experimental information in the 10-350 K temperature range document the complexity of commonly considered 'elementary' chemical reactions in a range relevant for modeling atmospheric and astrophysical environments. Perspectives are indicated for advances towards other types of transitions and to a global generality of processes of interest in applied chemical kinetics in biophysics and astrochemistry.
11:15 - 11:40 Piotr Żuchowski Ultracold Collisions of Li and Sr Atoms with Oxygen Molecules in Magnetic Fields: Prospects for Sympathetic CoolingStudies of cold molecules are one of the most exciting research areas in contemporary physics. In contrast to atoms, molecules offer a rich internal structure that can be exploited in precision spectroscopy and quantum technologies, such as clocks, simulators, and sensors. Here, I present studies on the ultracold collisions of Li and Sr atoms with oxygen molecules in a magnetic field, a typical condition in traps. The potential energy surfaces of these systems are very weakly anisotropic, which facilitates calculations and limits them to a low number of channels. While the low-energy collisions are extremely sensitive to the details of the potential, this is not the case for Li -- O2, since the reduced mass of the system is low, and the system supports a very low number of bound states. The prospects for sympathetic cooling of oxygen using these atoms are also discussed.
11:40 - 12:05 Alexandre Zanchet Reactivity of Ground and Excited Sulfur Cation with Molecular HydrogenSH+ is a widespread molecular ion in diffuse interstellar clouds, and has also been detected in emission toward the Orion Bar photodissociation region (PDR). In warm and dense PDRs, SH+ is predicted to be formed by exothermic reactions of S+(4S) with vibrationally excited H2 (v>1). The viability of this hypothesis has been confirmed by theoretical simulations which demonstrate that for vibrational level v=2 or higher of H2, the reaction exhibits high rate constants. To validate these theoretical predictions, the same potential energy surfaces will be employed to reproduce experimental cross sections on S+(4S)+H2 and S+(2D)+H2 reactions. Effects of spin-orbit interactions and of isotopic substitution of H2 on the S+(4S)+H2 reaction will be discussed, and the first results on S+(2D)+H2 reaction will be presented.
12:05 - 13:35 🍴 LunchSession 3 - Chair David Clary
13:35 - 14:10 Peter Saalfrank Quantum Dynamics of Simple Reactions in an EnvironmentQuantum reaction dynamics can be significantly altered when taking place in an environment. Here, two 'environments' will be considered, namely molecules embedded in a (Fabry-Pérot) cavity giving rise to strong coupling of vibrational degrees of freedom to cavity photons, and molecules at surfaces which are coupled to phonons. For the first example, using a so-called Pauli-Fierz Hamiltonian, the influence of cavity photons on reaction rates involving symmetric (inversion of ammonia) [1] and asymmetric double-well potentials (H-transfer) will be studied [2]. We demonstrate that by vibrational strong coupling, both rate deceleration and rate acceleration are possible. Strategies to actively control the reactivity of molecules in cavities will also be considered [2,3], as well as the validity of the cavity Born-Oppenheimer approximation frequently used in cavity reaction dynamics [4]. In the second case study, the phonon-assisted inversion of CO molecules at NaCl surfaces at low temperatures (~20 K) will be addressed. Specifically, the thermal reaction of an unusual 'O-down' isomer to the canonical 'C-down' form shows interesting features according to experiments [5]: The reaction is tunneling-dominated, with inversion rates exhibiting large, counterintuitive heavy-atom isotope effects and other unusual features such as small Arrhenius rate prefactors and sub-barrier activation energies. These effects can't be rationalized by standard scattering-type rate models such as transition state theory, the WKB model or instanton theory, but can be explained by a phonon-assisted, resonant-tunneling model (based on Fermi's Golden Rule) which involves 'tunneling gateway states' [5].
14:10 - 14:40 Marko Tomislav Cvitaš Tunneling Splittings in Molecules and Clusters Using Modified WKB Theory in Cartesian CoordinatesIn molecular systems with multiple equivalent symmetry-related minima separated by potential barriers, vibrational states can delocalize over the entire system via tunneling. This phenomenon manifests itself in the energy splittings of the delocalized states in vibrational spectra. Tunneling splittings vary over many orders of magnitude depending on the type of rearrangement involved and modal excitations of the system. Calculation of tunneling splittings using variational methods for molecules larger than a few atoms is computationally prohibitive. We present a semiclassical theory for calculating tunneling spectra that works in full dimensionality and is scalable to systems of high dimensionality. Our approach is based on a modified WKB theory on the minimum action paths connecting the symmetry-related minima [1-3]. We show that our approach is equivalent to the instanton theory for tunneling splittings of the ground vibrational state and that it gives an exact wavefunction in a harmonic potential for the ground and the first excited state. We test it on the vinyl radical molecule [4] and apply it to calculate tunneling patterns in a number of water clusters in the ground and vibrationally excited states. We also discuss an extension to calculate the splittings in rotationally excited states with an application to the HO2 molecule and future directions with a view of calculating tunneling rates in molecules at low temperatures.
14:40 - 15:05 Tiangang Yang Quantum State-Selected Reaction Dynamics at Low TemperaturesQuantum state-selected scattering at low temperatures is crucial for understanding molecular reaction dynamics and the chemistry of interstellar space. Central to these experimental studies are techniques that lower the temperature of reactants and precisely select their quantum states. Recently, we have integrated a laser-cooled ion trap system with a high-resolution time-of-flight mass spectrometer (TOF-MS) and fluorescence imaging to explore reaction dynamics. This apparatus further includes a stimulated Raman pumping (SRP) system for molecular state selection. In this presentation, I will discuss the latest research advancements from this project.

QRS 2024 Powerhouse
We are proud to feature Alpine Dynamics GmbH, a company based in Switzerland, as the principal financial steward for QRS 2024. Alpine Dynamics goes beyond traditional sponsorship and partnership roles by exclusively executing all financial transactions associated with the event. We extend our deepest gratitude to Alpine Dynamics for their critical role and extensive support in the financial management of QRS 2024.
QRS 2024 Sponsors
QRS 2024 Partners

Venue
The forthcoming QRS 2024 event is set to take place at the Yıldız Campus, which is situated within the historic bounds of the Yıldız Palace complex. This campus represents the university's original establishment, taking its name directly from the palace; "Yıldız" translates to "star" in English. The image presented below showcases the auditorium designated for QRS 2024, accompanied by a selection of photographs from the Yıldız campus.






Gala Dinner
The QRS 2024 Gala Dinner is planned for Thursday, June 27, 2024, at the Hisar Restaurant in Istanbul. Located near the famous bridge that connects the two continents Istanbul is on, the Yıldız Hisar Restaurant is only 7 kilometers (4.35 miles) away from the QRS 2024 venue. There will be a free ride provided by QRS 2024, making it easy to get there. Not only does the restaurant offer stunning views of the Bosphorus and the bridge, but the menu is also exceptional, promising a delightful culinary experience. The photos showcasing the restaurant's breathtaking scenery have not been edited, ensuring you will enjoy these beautiful sights just as they are. QRS 2024 Gala dinner is set to be an unforgettable evening with both delicious food and spectacular views.






Lunches & Coffee Breaks
At QRS 2024, we are not only setting the stage for pioneering scientific discussions but also for an exceptional gastronomic experience.
As organizers, we firmly believe that fine dining is an essential part of the meeting experience. We are excited to show that
Turkish cuisine is much more than just
doner kebab.
Be ready to explore the rich mosaic of flavors that Turkish gastronomy has to offer.
For five days, we will be collaborating with a prestigious catering company to deliver the authentic tastes of Turkish cuisine.
During coffee breaks, enjoy the indispensable sweets from Turkish patisseries. Make sure to check our website's dining section;
we will soon unveil the daily menus designed to enhance your QRS 2024 experience. QRS participants will meet extraordinary dining
in the heart of Istanbul!

Travel Arrangements
We understand the importance of seamless planning for your trip to QRS 2024, especially for our international participants coming from various countries. Here, we aim to provide you with all the necessary information to make your journey as smooth and enjoyable as possible.

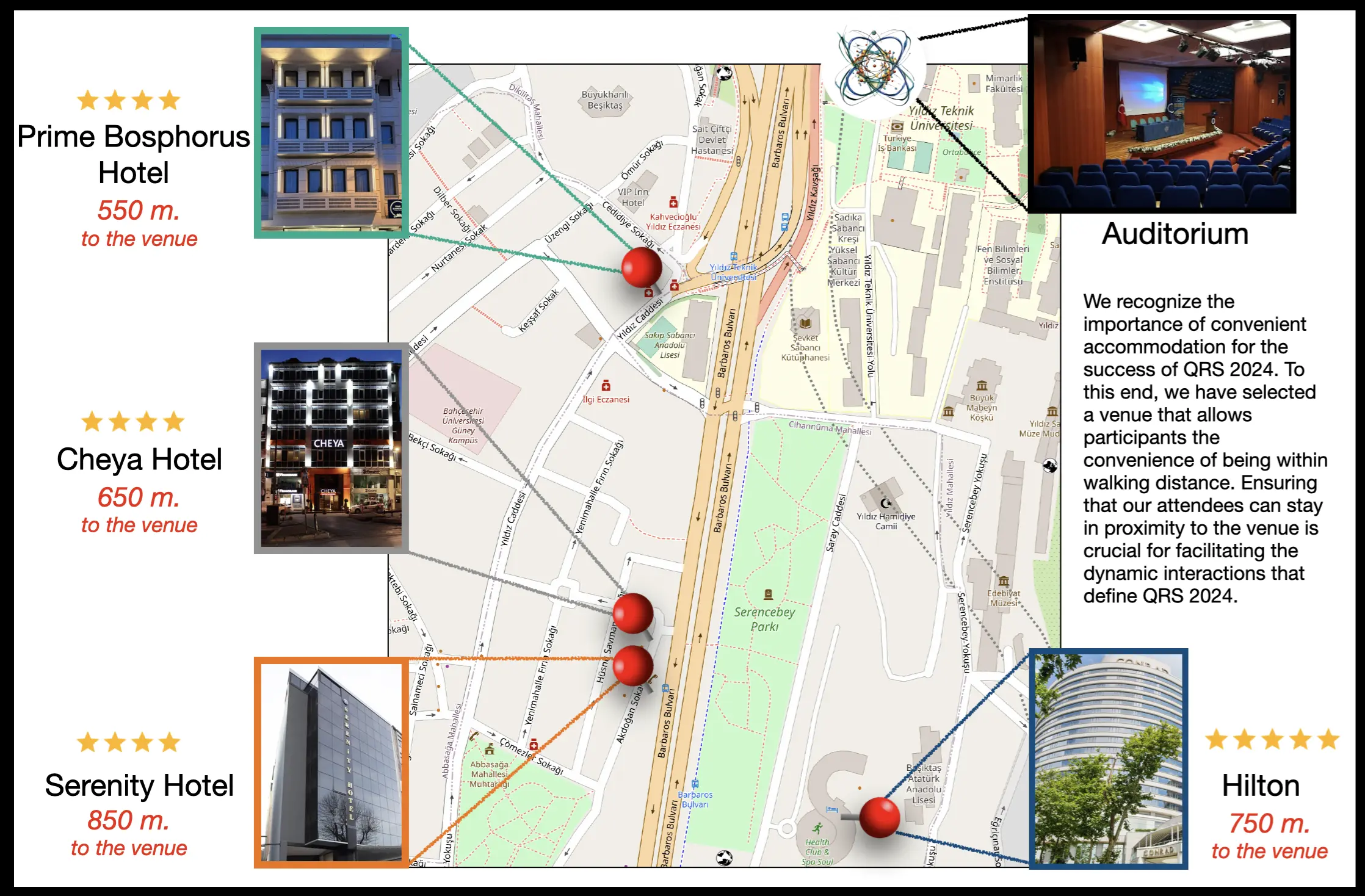



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
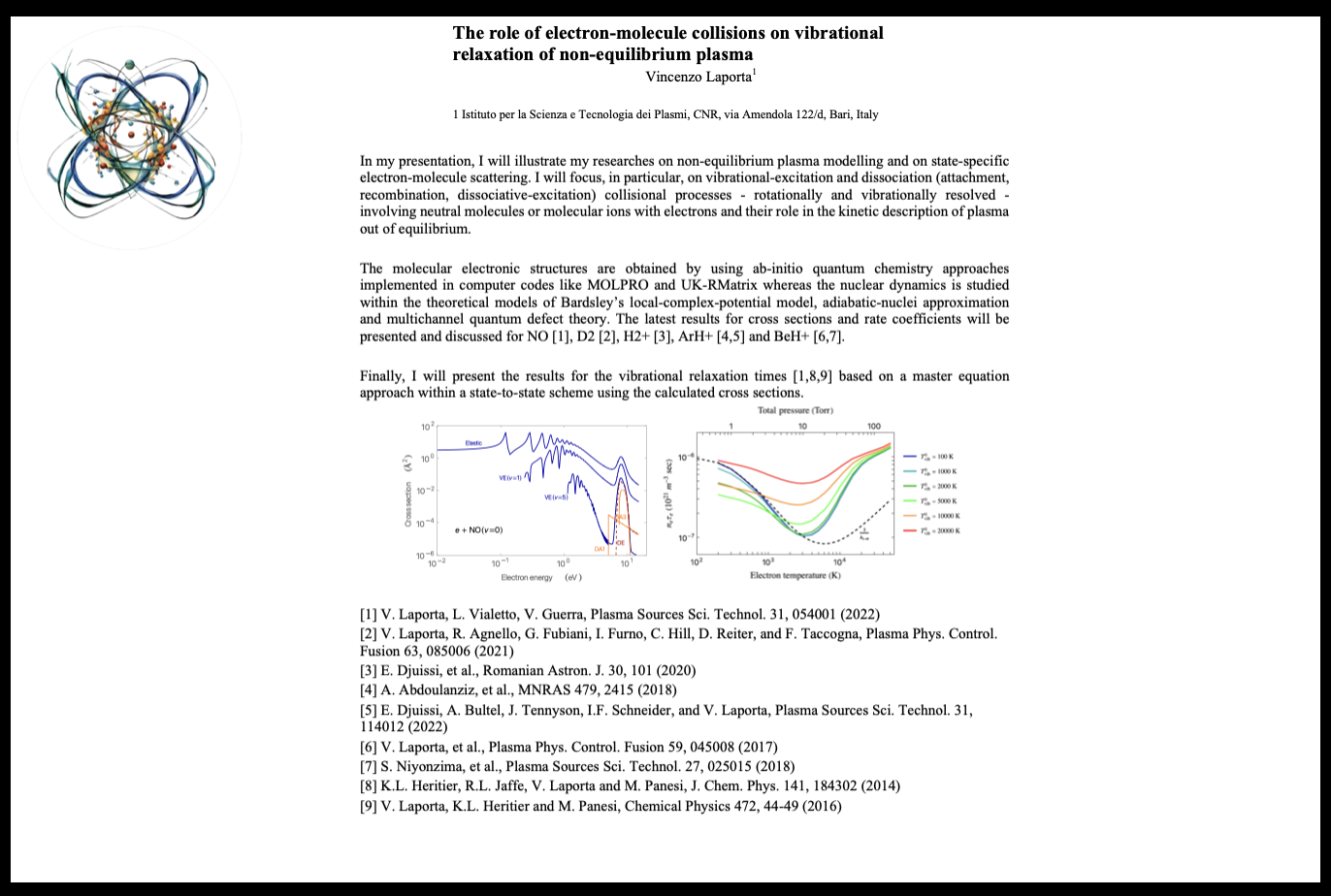{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
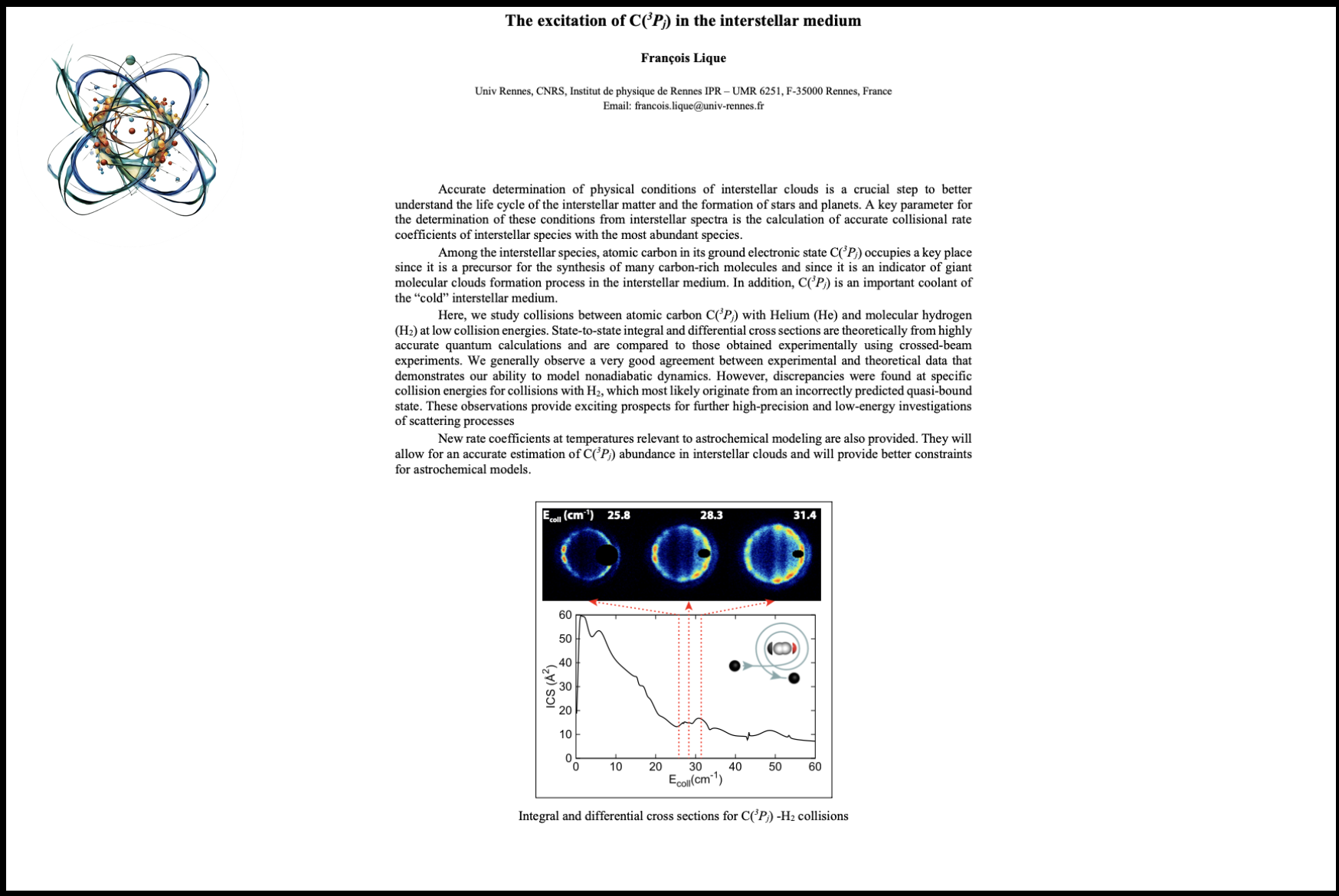{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
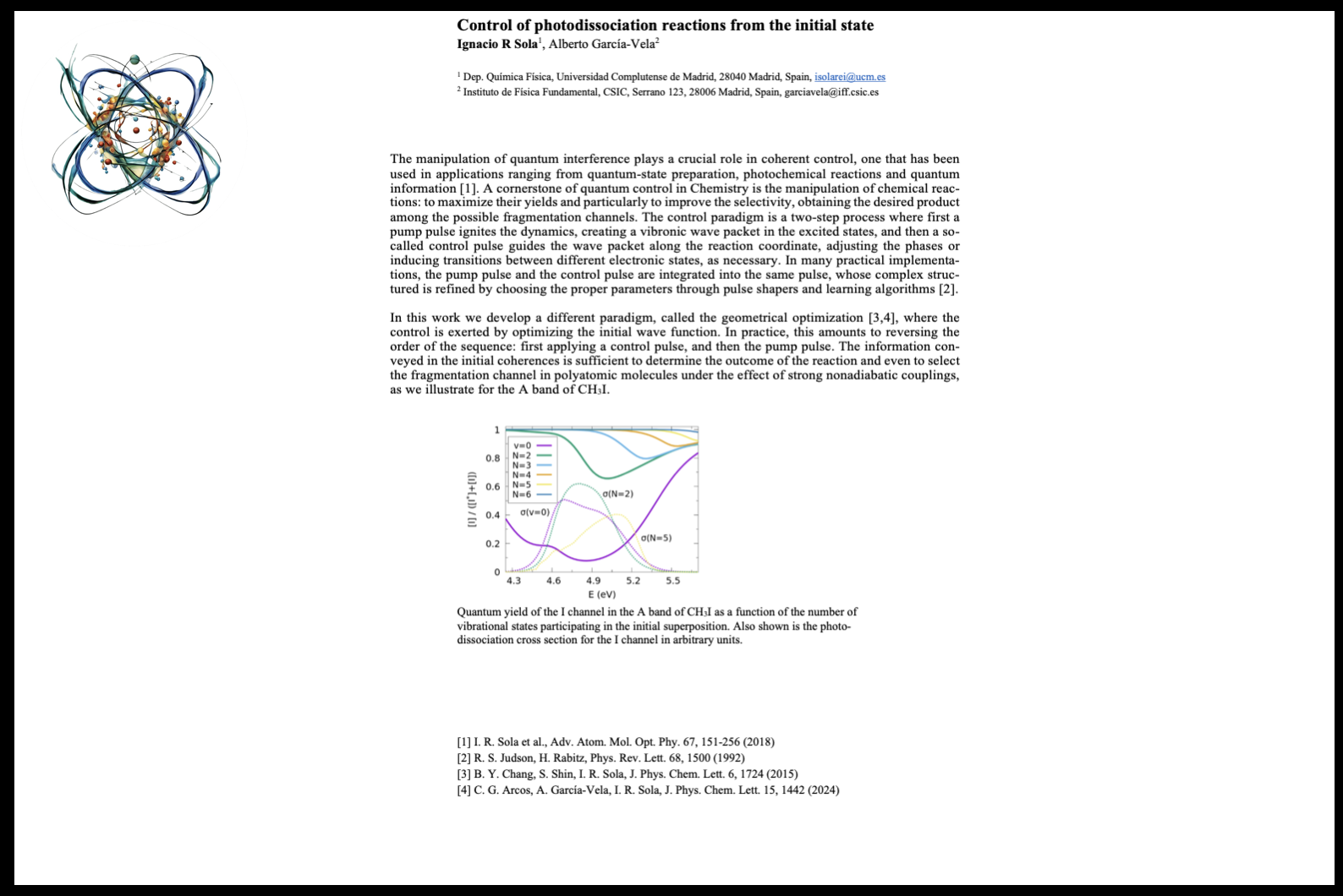{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
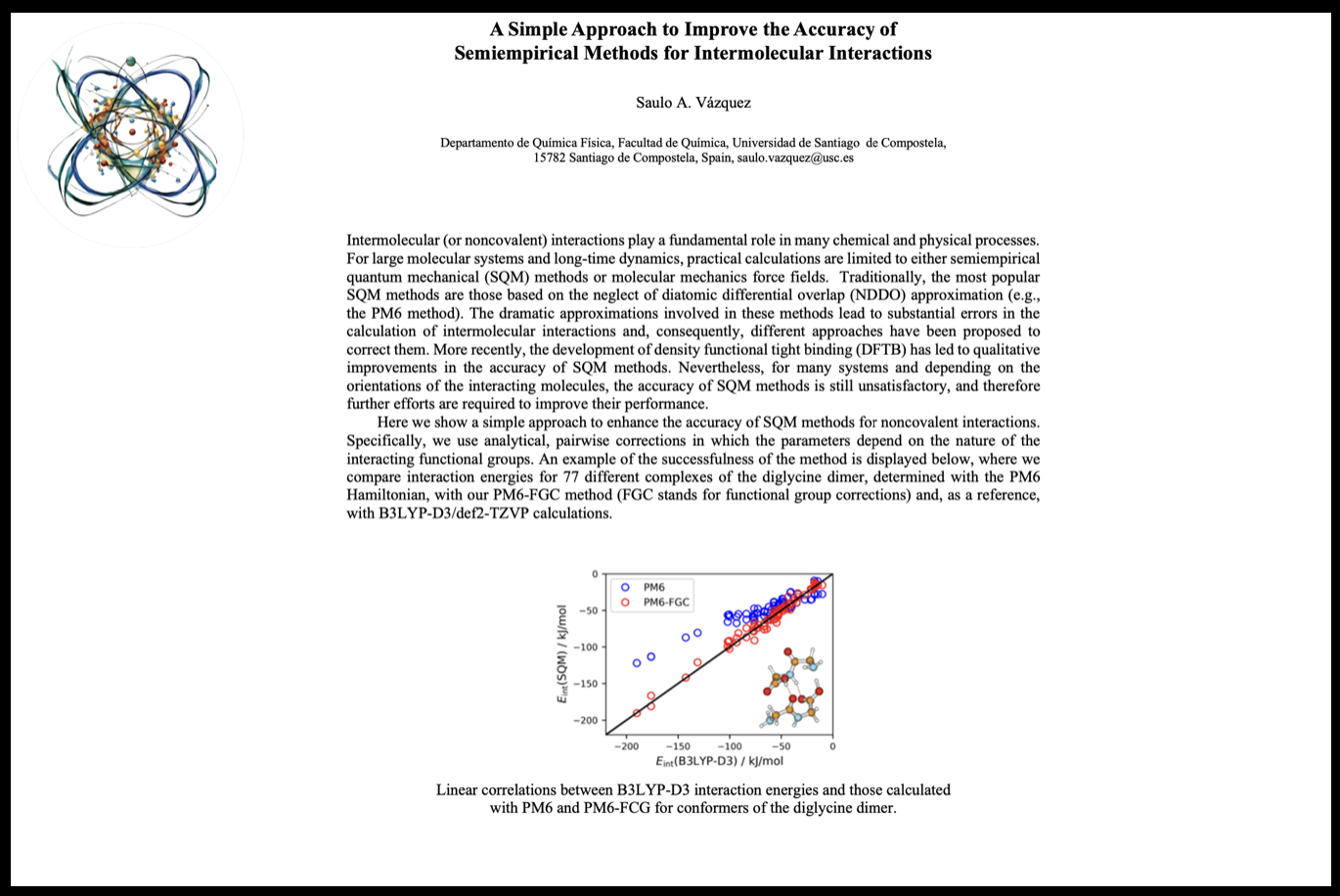{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Registration
Please click here to register for QRS 2024.
| Registration Fee | Full | Reduced (Students) | Accompanying Person |
|---|---|---|---|
| up to April 22nd, 2024 | € 625 | € 365 | € 225 |
| up to May 14th, 2024 | € 715 | € 415 | € 225 |
Please note that the workshop will be held in-person, without any online or recorded video talk
Registration fee includes:
🍸 Welcome Reception All participants gather for a delightful start at the enchanting welcome reception
🍽️ Gala Dinner Delicious food in a delightful atmosphere, overlooking the magnificent Istanbul Bosphorus
🍹 Cocktail during Poster Presentations Refreshments and snacks along with networking at Poster Session
⛵ Cocktail on a Boat: Bosphorus Tour 🍹 🐟 Scenic views await: Hagia Sophia, Maiden Tower, Topkapı Palace, Galata Tower, Dolmabahçe Palace, Çırağan, Ortaköy Mosque, Bosphorus Bridge, Beylerbeyi Palace, and Üsküdar during the Bosphorus tour
🍴 Lunch Enjoy delicious lunch options every day of QRS 2024
☕ Coffee Break Refreshing coffee breaks with various Turkish pastries throughout the event
🧳 Workshop materials Book of Abstracts (astonishingly, in its tangible form!), certificate of participation, QRS 2024 bag
Accompanying person fee includes: 🍸 Welcome Reception 🍽️ Gala dinner 🍹🚢 🐟 Cocktail on a Boat 🍴 Lunches
Upload Your Presentation to the QRS Server
During our recent meeting with the auditorium staff at the conference center where QRS 2024 will be held, they provided us with some valuable
recommendations regarding the efficient running of the scientific program. Based on their experiences at other conferences and workshops,
they have observed that time can be lost when different personal computers are connected to the projector during each speaker's presentation.
To ensure a smoother program flow, they emphasized the importance of speakers uploading their presentations (in PDF, PowerPoint, or Keynote format) in advance.
To facilitate this, we have set up a server where you can upload your presentation.
Your uploaded presentation will not be shared with any third parties.
It will be deleted from the server after your presentation.
We recommend uploading your presentation to our system via our website no later than 24 hours before your scheduled talk.
Thank you for your cooperation.
Poster Guidelines
Poster Size: Posters should be prepared in A0 size (84.1 cm x 118.9 cm or 33.1 inches x 46.8 inches).
Orientation: Posters must be in portrait orientation.
Title: The title should be bold and clearly visible at the top of the poster. Include the names of the authors and their affiliations.
Introduction: Provide a brief background and the objectives of your research.
Methods: Describe the methods used in your research concisely.
Results: Present your findings with clear figures, tables, and graphs.
Discussion: Summarize the implications of your results.
Conclusion: Provide a brief summary of your key findings and their significance.
References: Include any references cited in your poster.
Font Size: Ensure that all text is easily readable from a distance of 1-2 meters. The recommended font size for the title is at least 56 points, section headers at least 32 points, and body text at least 14 points.
Fonts: Use a clear and readable font such as Arial, Calibri, or Times New Roman.
Colors: Use contrasting colors for text and background to enhance readability.
Graphics: Use high-resolution images, charts, and graphs. Ensure they are clear and legible. Captions: Provide clear and concise captions for all visuals.
Display Time: Posters should be set up at least one hour before the poster session begins and remain displayed for the entire session. Mounting: Mounting materials will be provided at the venue.
At least one author should be present at the poster during the designated poster session time to discuss the work and answer questions.
QRS 2024's logo should be placed at the top left corner of all posters. Please click here to download the QRS 2024 workshop logo.
{kind=link}
While you might not be carrying as much luggage as the traveler in this image, a poster tube can still feel like an extra burden. We understand the challenges of transporting poster tubes, especially when flying. For your convenience, there is a printing office located just 1 km from the venue and approximately 300 meters from the QRS-designated hotels, offering A0 poster printing services for around 13 Euros. If you wish to use their services, we can assist in facilitating communication with the printing company. You can send your poster to them by e-mail and collect it upon arrival in Istanbul. This option is entirely at your discretion.
